

A Systematic Protein Turnover Map for Decoding Protein Degradation
- PMID: 33176155
- PMCID: PMC7745259
- DOI: 10.1016/j.celrep.2020.108378
A Systematic Protein Turnover Map for Decoding Protein Degradation
Abstract
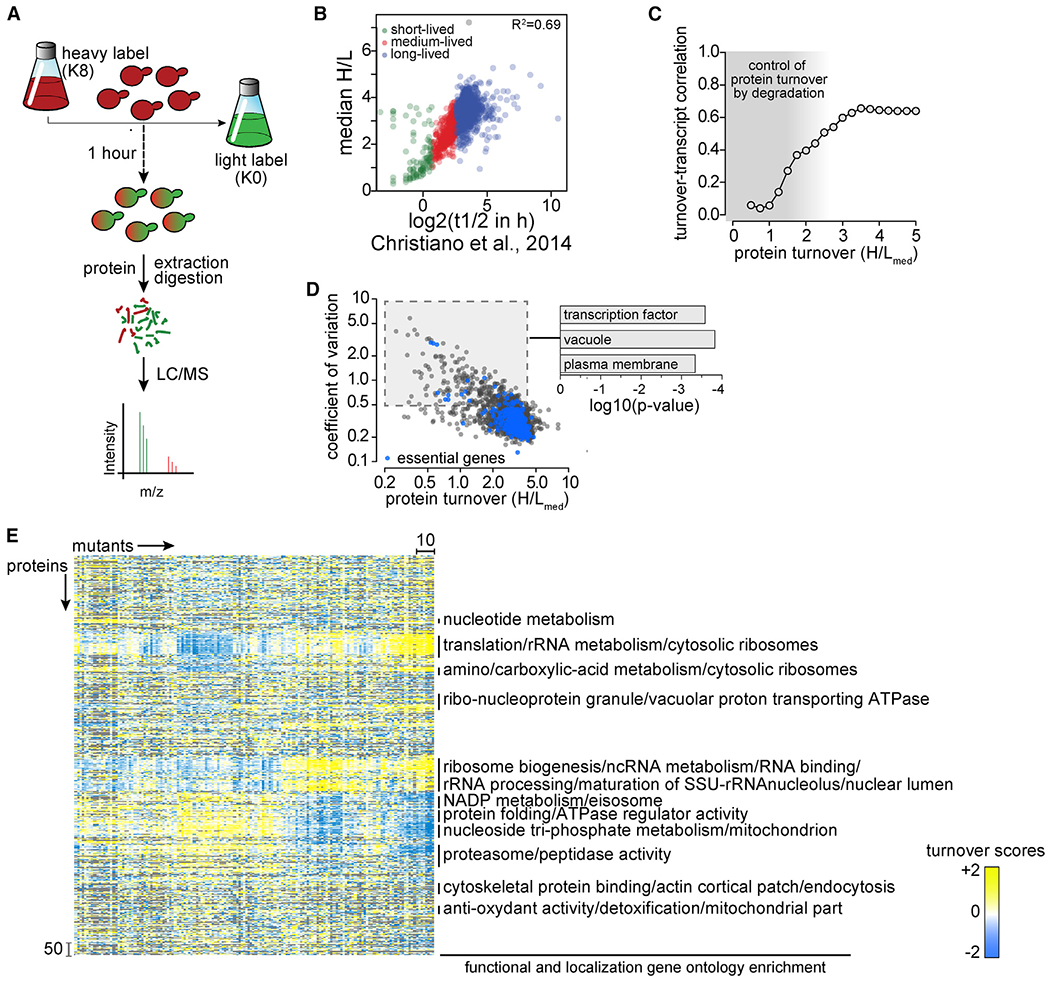
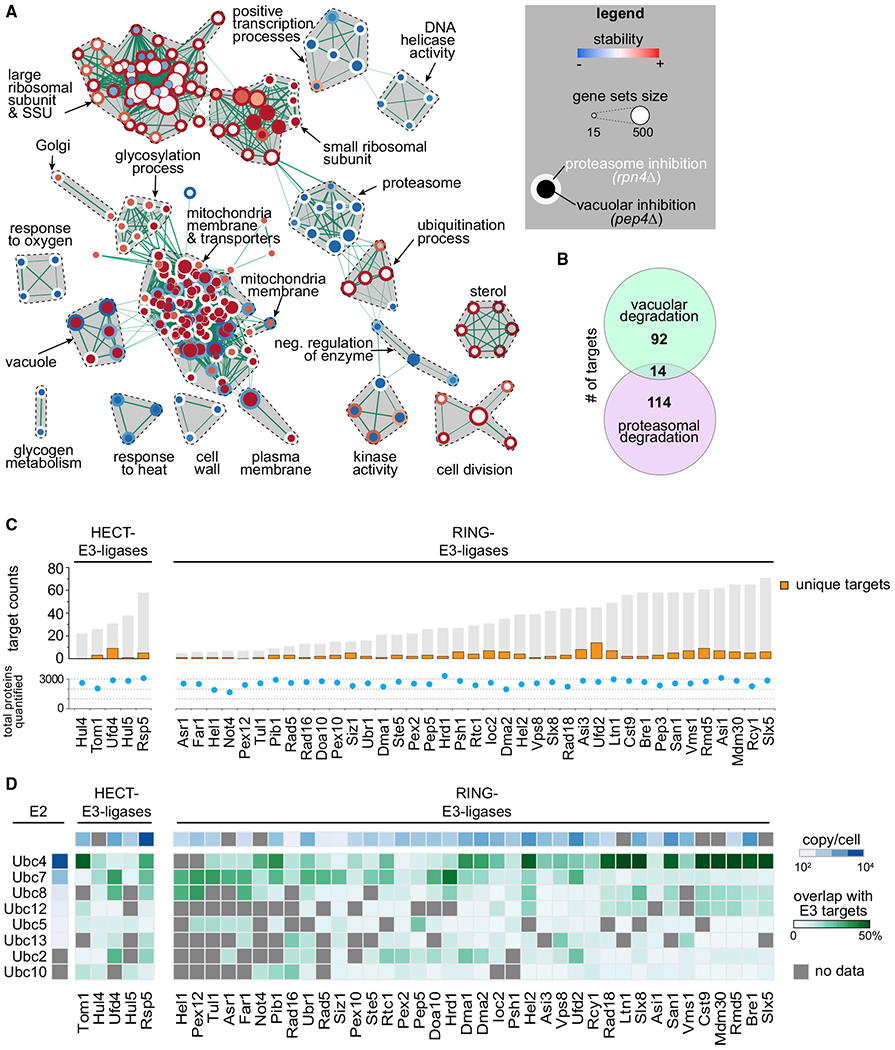
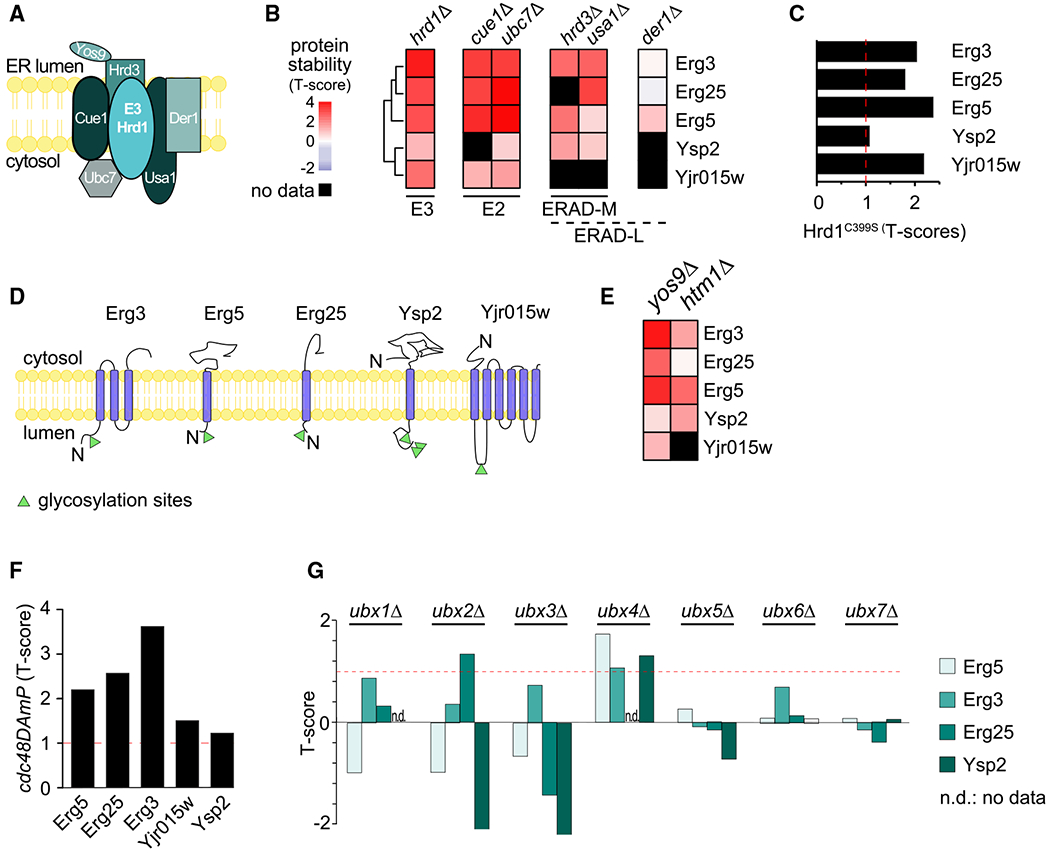
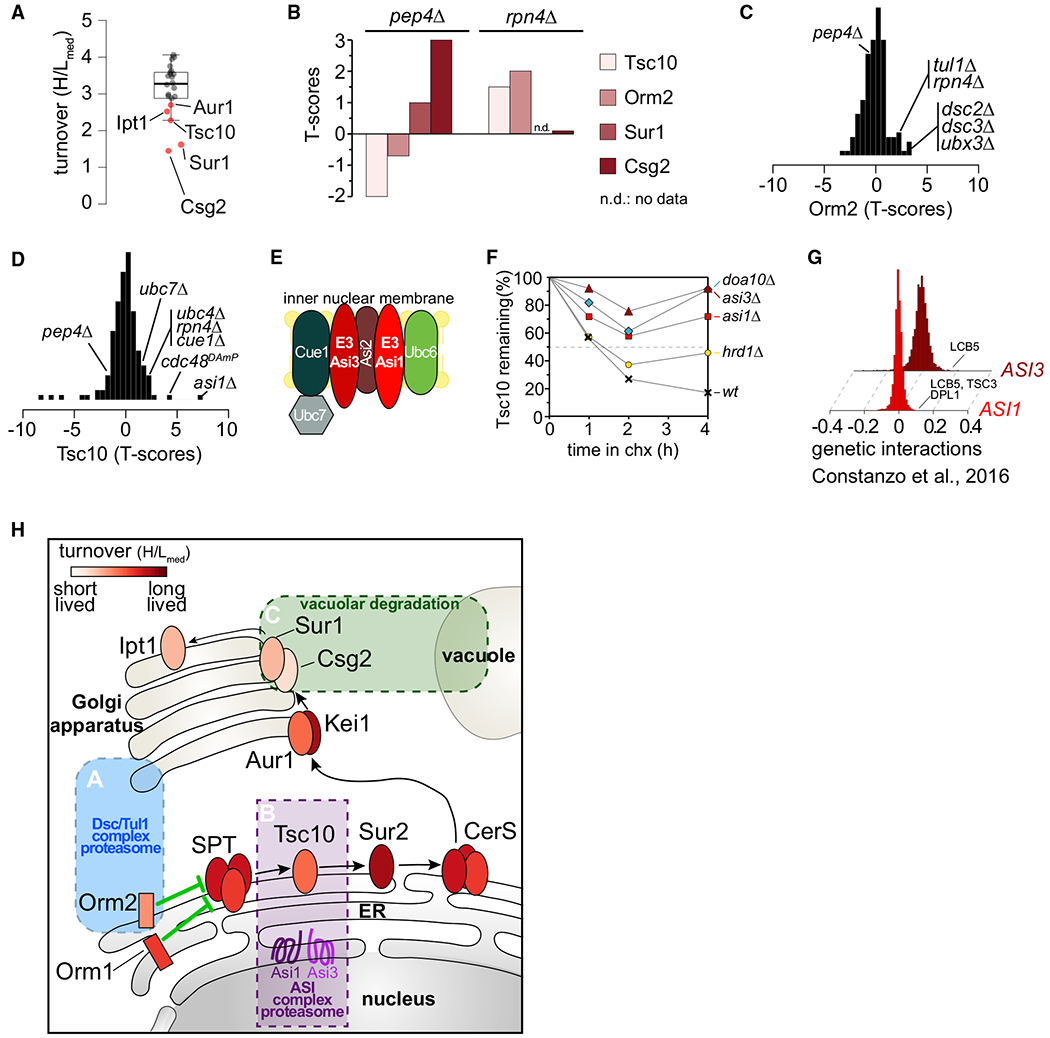
Protein degradation is mediated by an expansive and complex network of protein modification and degradation enzymes. Matching degradation enzymes with their targets and determining globally which proteins are degraded by the proteasome or lysosome/vacuole have been a major challenge. Furthermore, an integrated view of protein degradation for cellular pathways has been lacking. Here, we present an analytical platform that combines systematic gene deletions with quantitative measures of protein turnover to deconvolve protein degradation pathways for Saccharomyces cerevisiae. The resulting turnover map (T-MAP) reveals target candidates of nearly all E2 and E3 ubiquitin ligases and identifies the primary degradation routes for most proteins. We further mined this T-MAP to identify new substrates of ER-associated degradation (ERAD) involved in sterol biosynthesis and to uncover regulatory nodes for sphingolipid biosynthesis. The T-MAP approach should be broadly applicable to the study of other cellular processes, including mammalian systems.
Keywords: E2; E3 ligases; ERAD; SILAC; mass spectrometry; proteasome; protein turnover; proteomics; ubiquitin.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Arrigo AP, Tanaka K, Goldberg AL, and Welch WJ (1988). Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome). Nature 331, 192–194. - PubMed
-
- Arthington BA, Bennett LG, Skatrud PL, Guynn CJ, Barbuch RJ, Ulbright CE, and Bard M (1991). Cloning, disruption and sequence of the gene encoding yeast C-5 sterol desaturase. Gene 102, 39–44. - PubMed
-
- Beeler T, Bacikova D, Gable K, Hopkins L, Johnson C, Slife H, and Dunn T (1998). The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2Delta mutant. J. Biol. Chem 273, 30688–30694. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
