

Progressive Cactus is a multiple-genome aligner for the thousand-genome era
- PMID: 33177663
- PMCID: PMC7673649
- DOI: 10.1038/s41586-020-2871-y
Progressive Cactus is a multiple-genome aligner for the thousand-genome era
Abstract
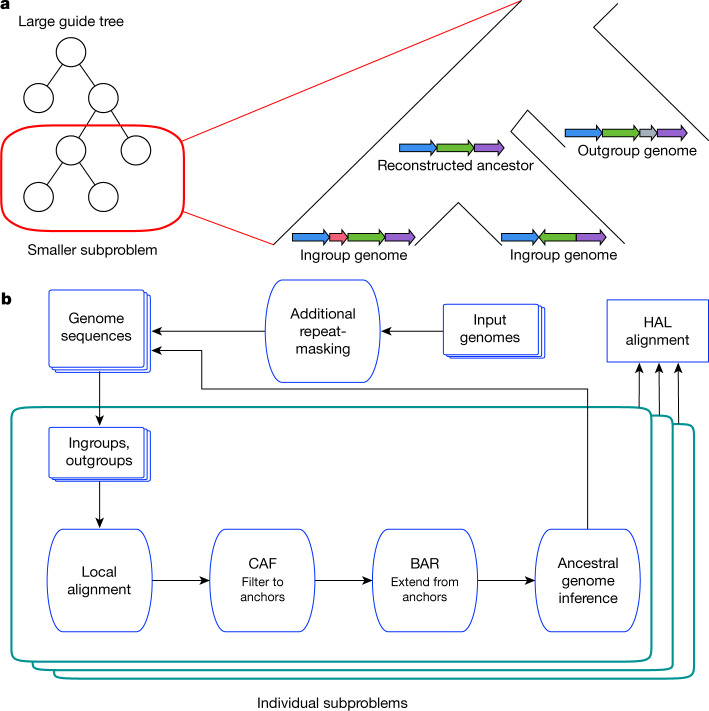
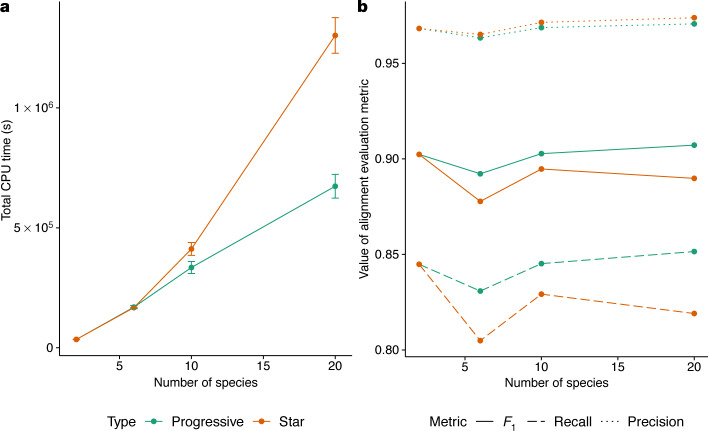
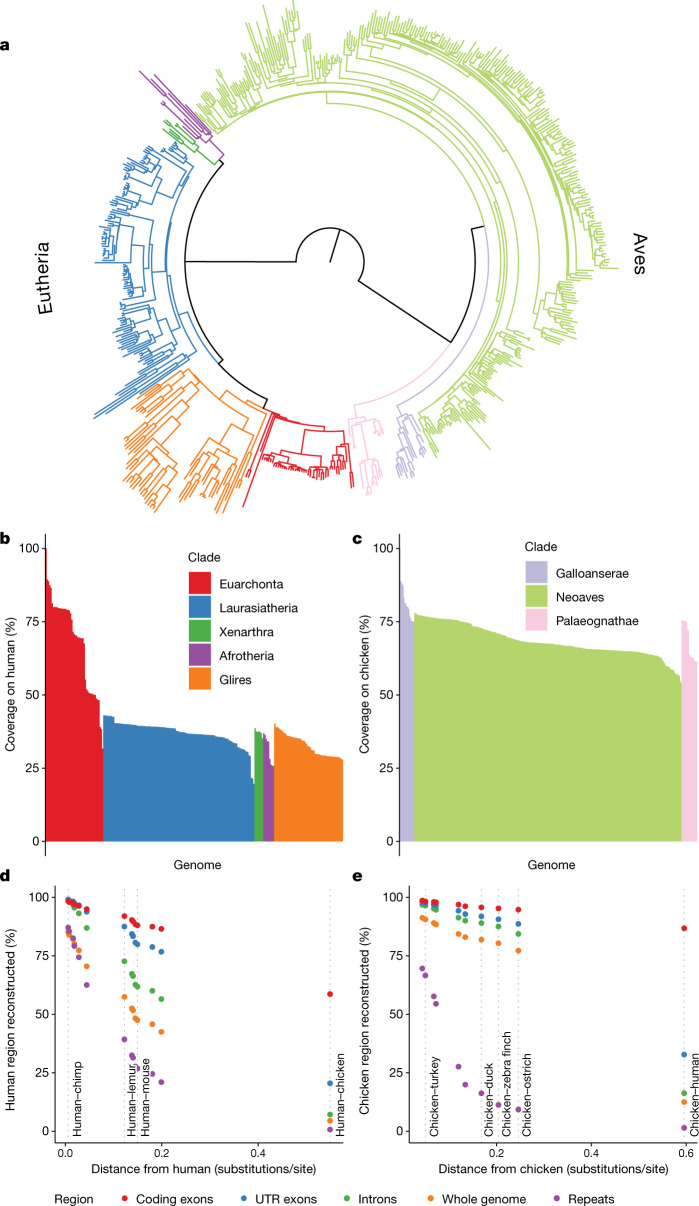
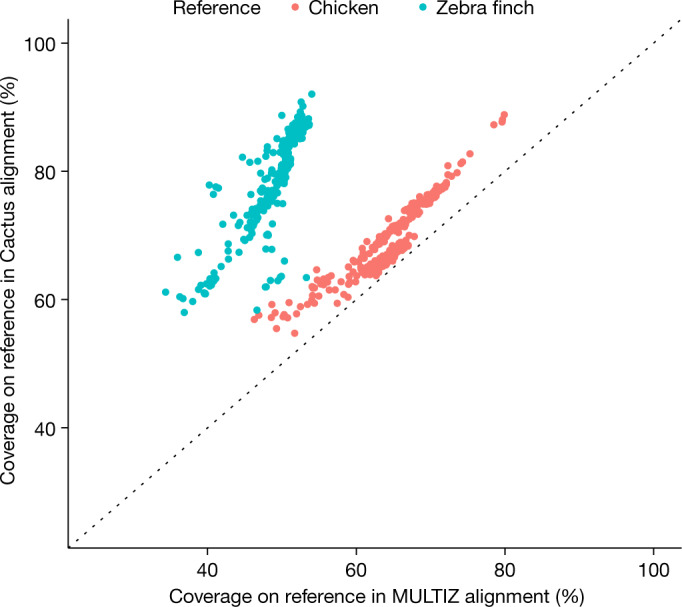
New genome assemblies have been arriving at a rapidly increasing pace, thanks to decreases in sequencing costs and improvements in third-generation sequencing technologies1-3. For example, the number of vertebrate genome assemblies currently in the NCBI (National Center for Biotechnology Information) database4 increased by more than 50% to 1,485 assemblies in the year from July 2018 to July 2019. In addition to this influx of assemblies from different species, new human de novo assemblies5 are being produced, which enable the analysis of not only small polymorphisms, but also complex, large-scale structural differences between human individuals and haplotypes. This coming era and its unprecedented amount of data offer the opportunity to uncover many insights into genome evolution but also present challenges in how to adapt current analysis methods to meet the increased scale. Cactus6, a reference-free multiple genome alignment program, has been shown to be highly accurate, but the existing implementation scales poorly with increasing numbers of genomes, and struggles in regions of highly duplicated sequences. Here we describe progressive extensions to Cactus to create Progressive Cactus, which enables the reference-free alignment of tens to thousands of large vertebrate genomes while maintaining high alignment quality. We describe results from an alignment of more than 600 amniote genomes, which is to our knowledge the largest multiple vertebrate genome alignment created so far.
Conflict of interest statement
The authors declare no competing interests.
Figures
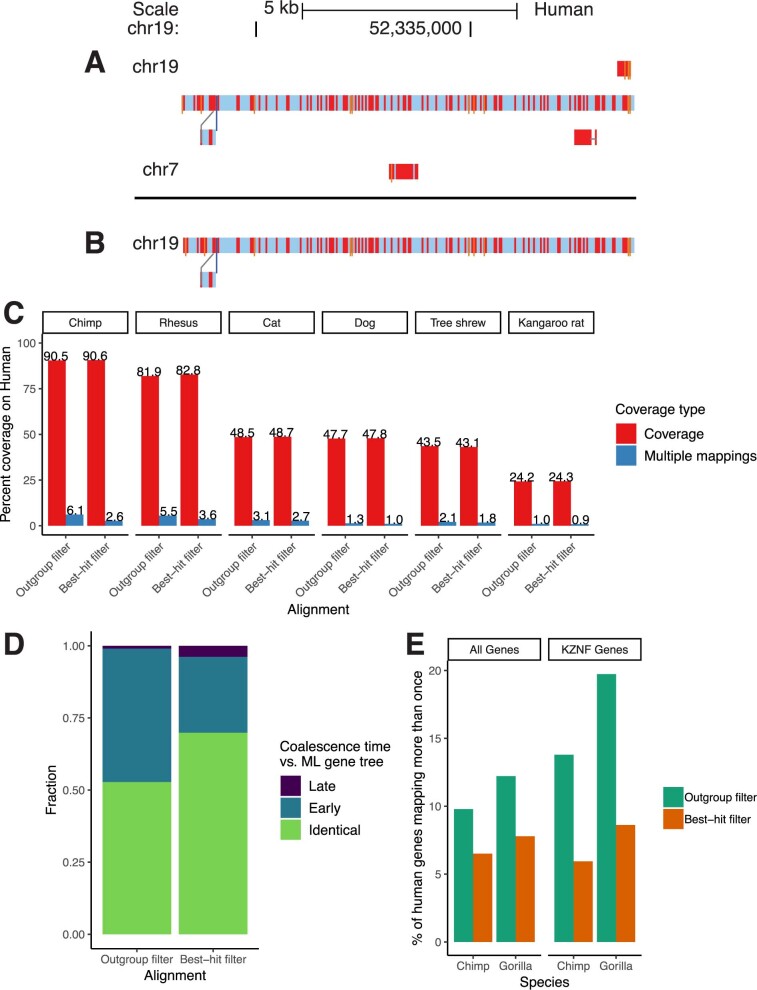
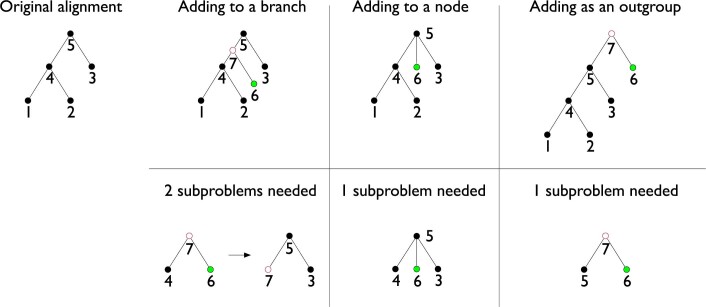
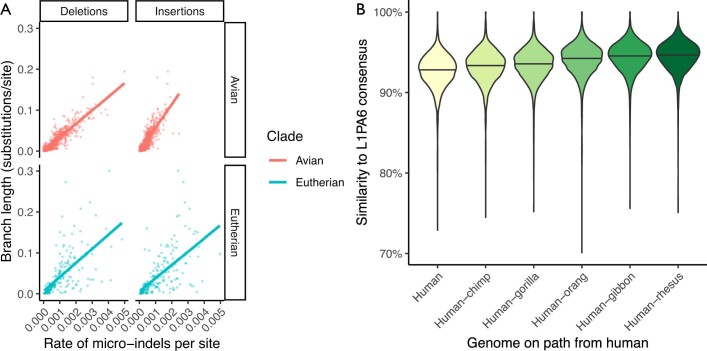
Comment in
-
Scaling up multiple-genome alignments.Nat Methods. 2021 Jan;18(1):33. doi: 10.1038/s41592-020-01045-8. Nat Methods. 2021. PMID: 33408401
References
-
- Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. - PubMed
Publication types
MeSH terms
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- U01 HL137183/HL/NHLBI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- 5T32HG008345-04/NH/NIH HHS/United States
- R01 HG008742/HG/NHGRI NIH HHS/United States
- U41 HG007234/HG/NHGRI NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- T32 HG008345/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- R01 HG009737/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U54 HG007990/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
