

Transcriptome and translatome co-evolution in mammals
- PMID: 33177713
- PMCID: PMC7116861
- DOI: 10.1038/s41586-020-2899-z
Transcriptome and translatome co-evolution in mammals
Abstract
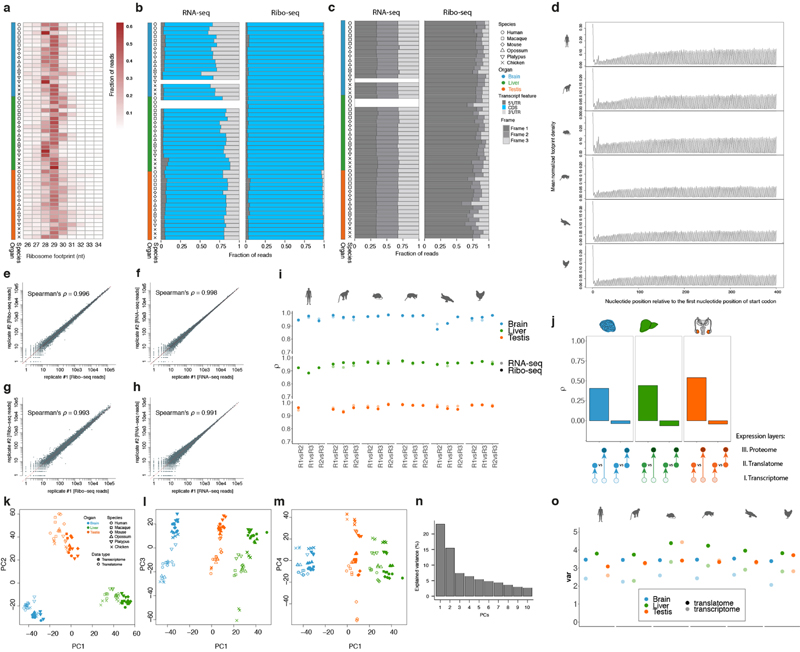
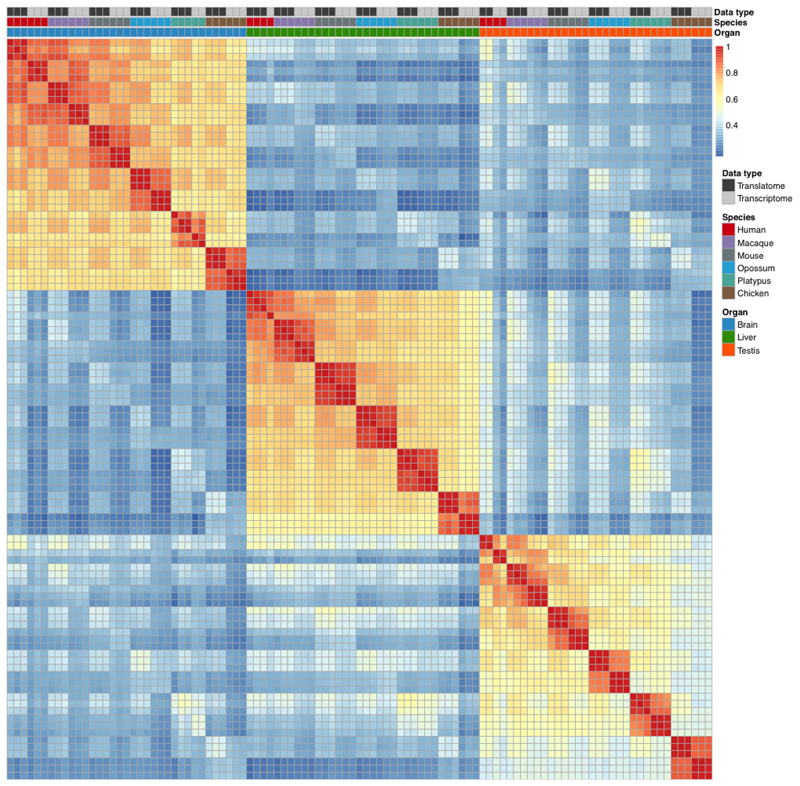
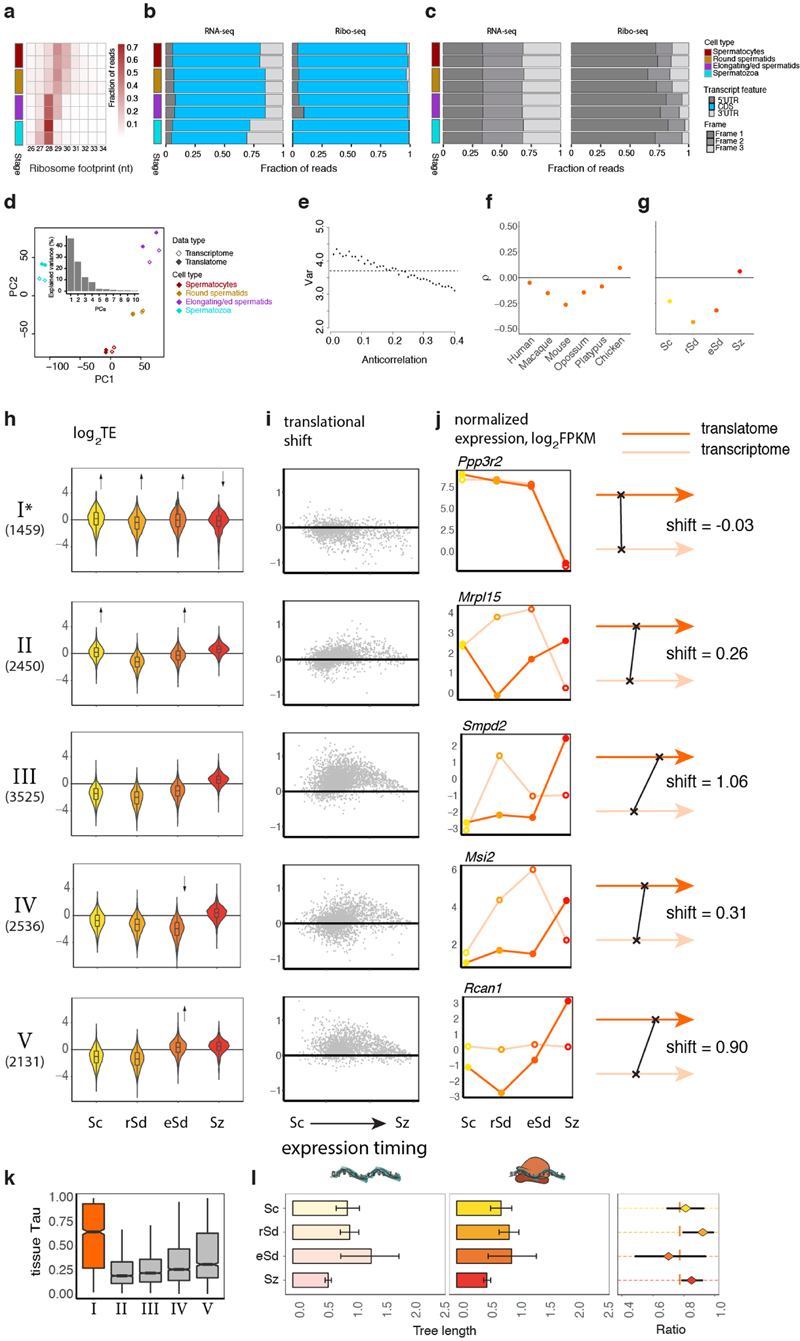
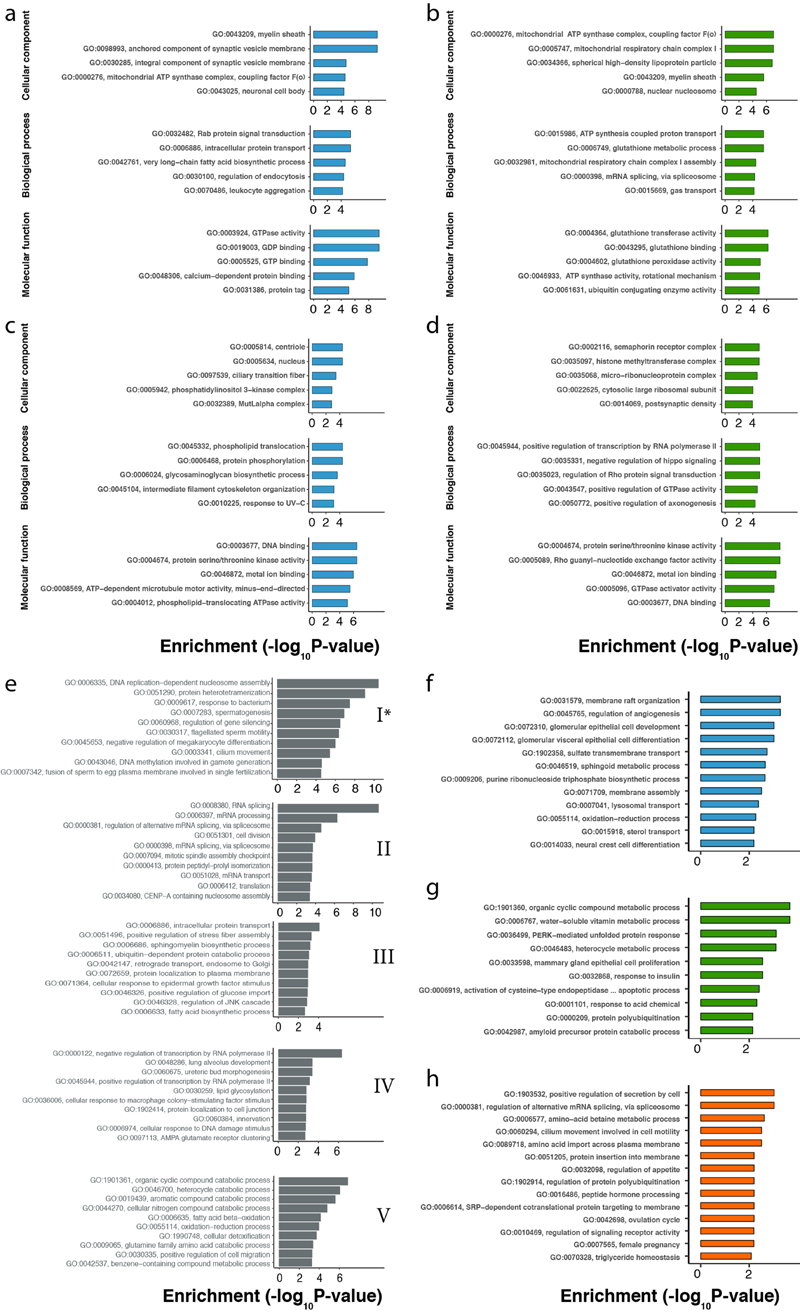
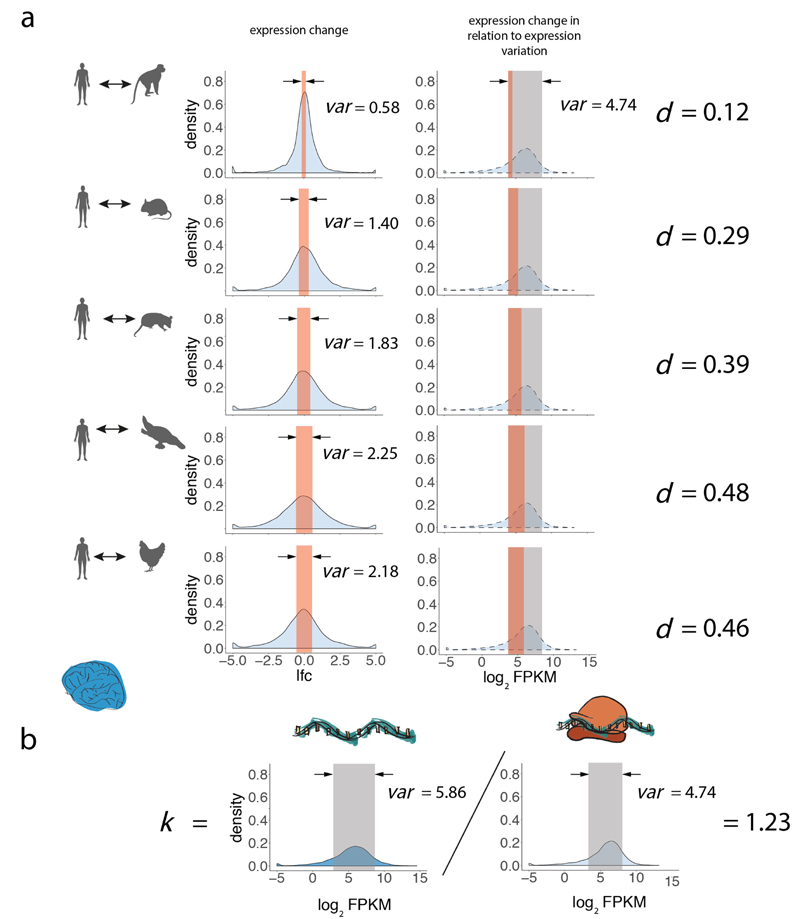
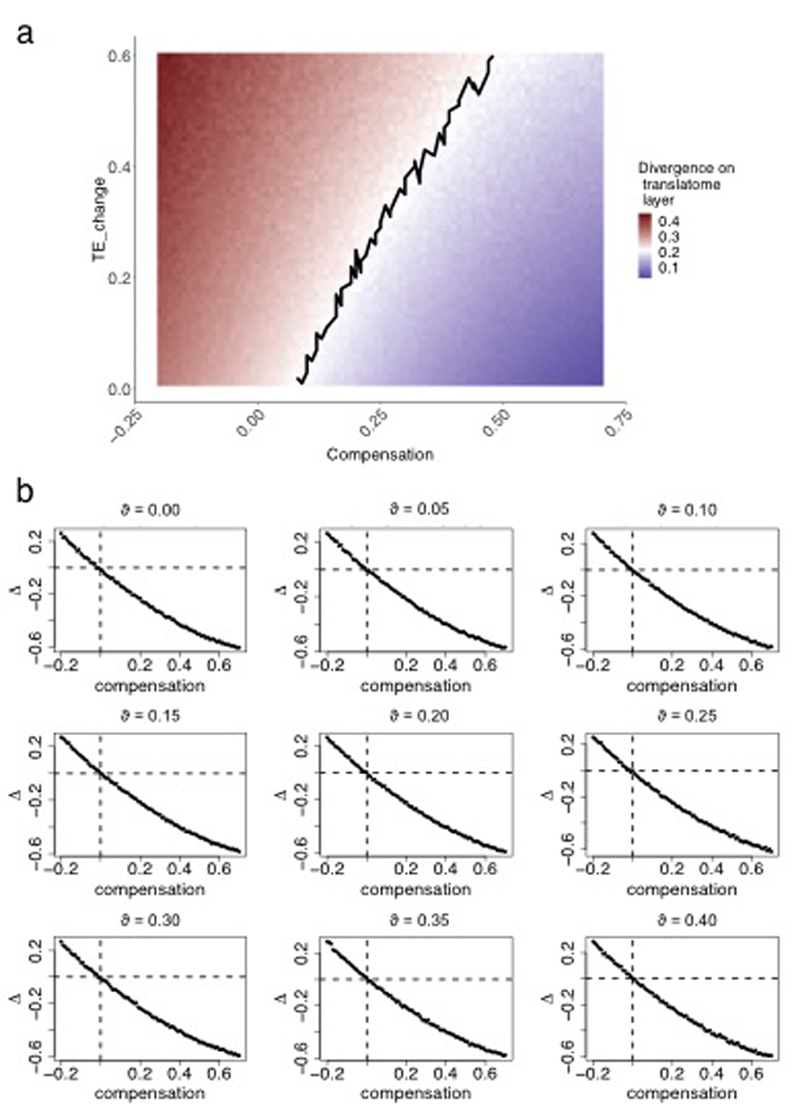
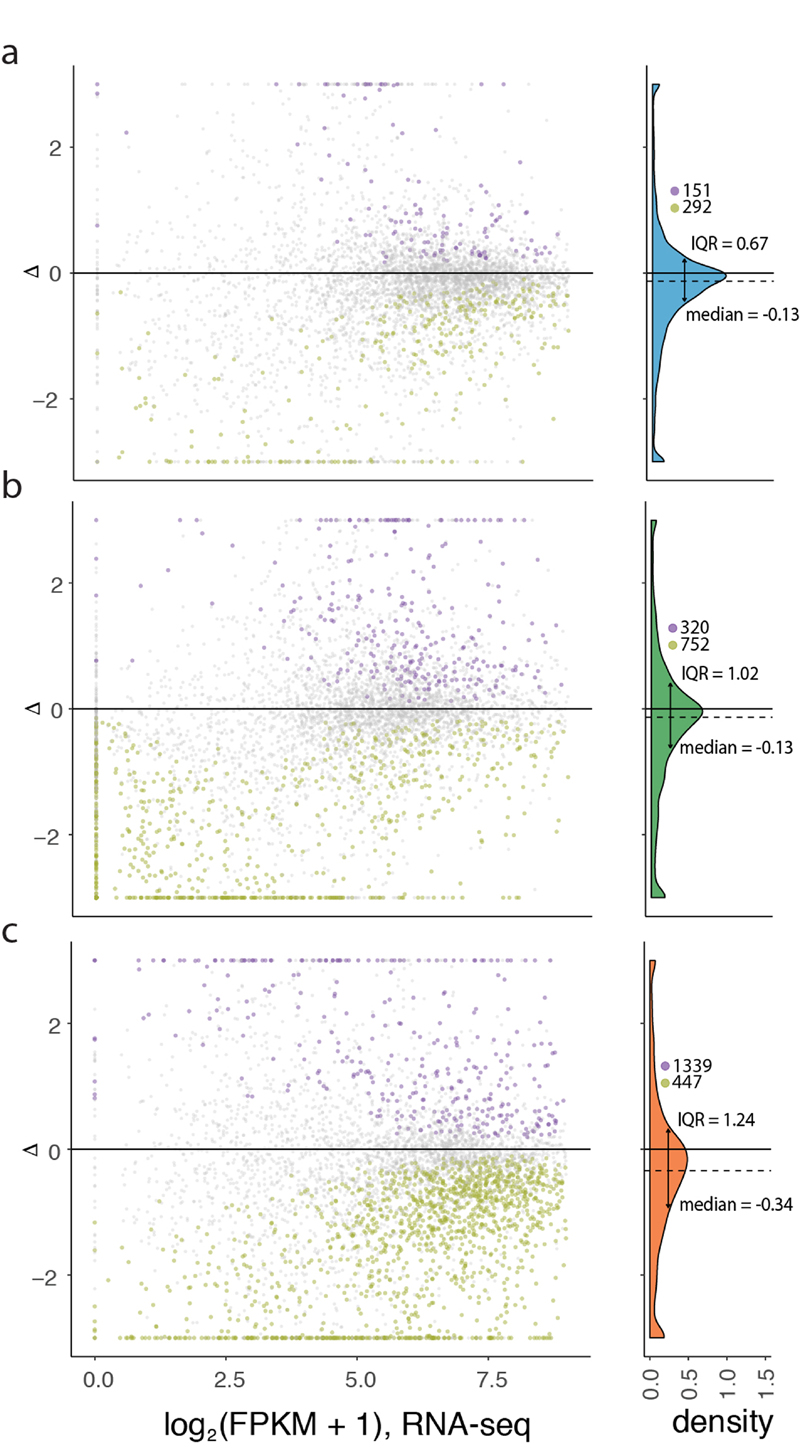
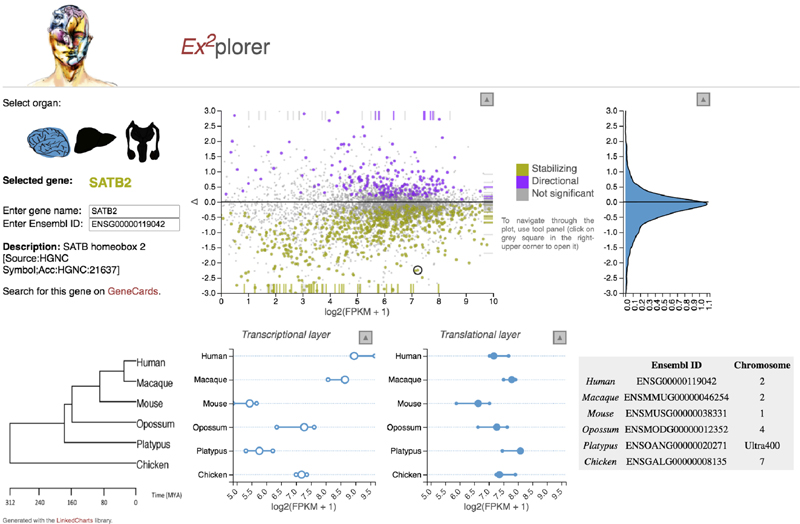
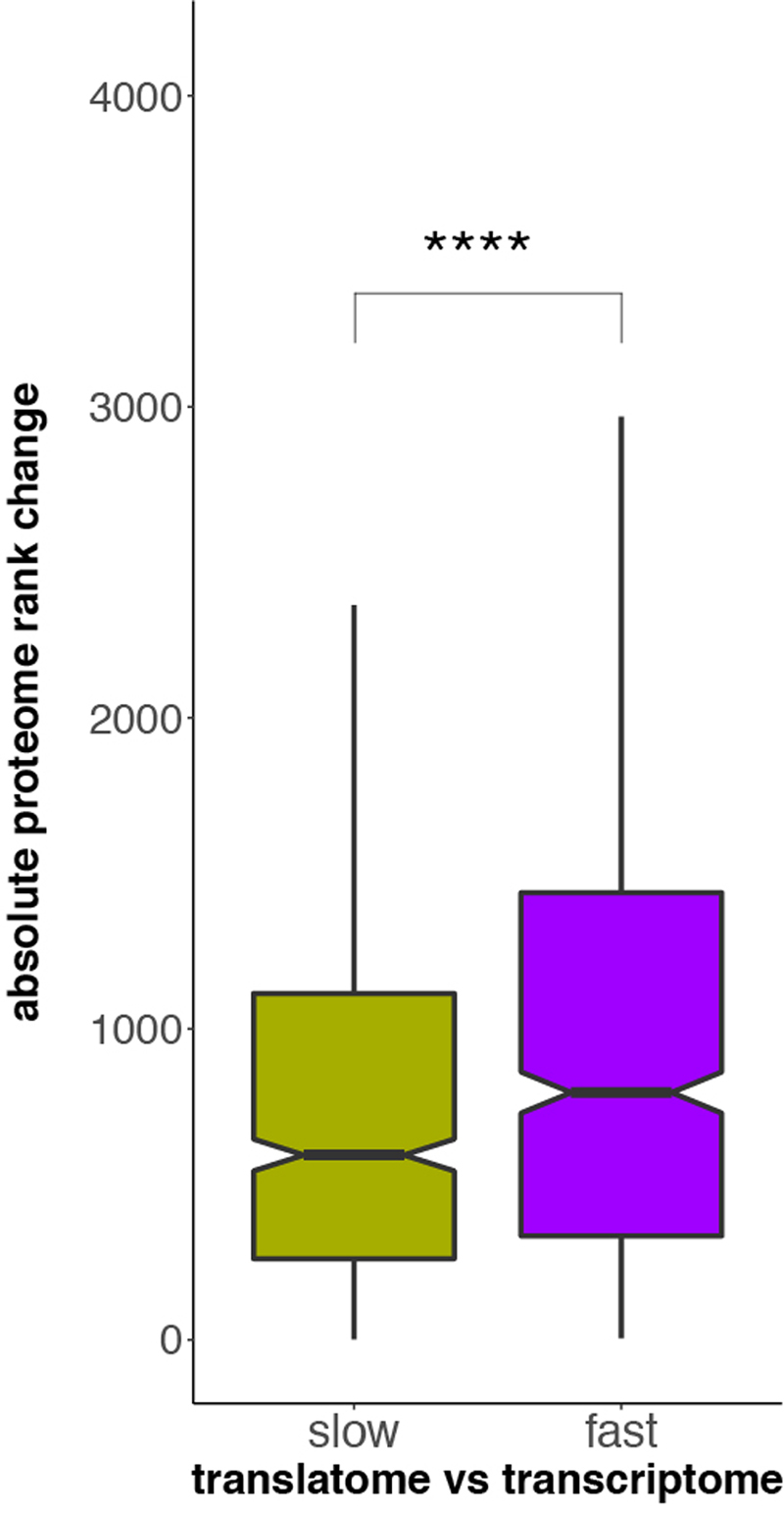
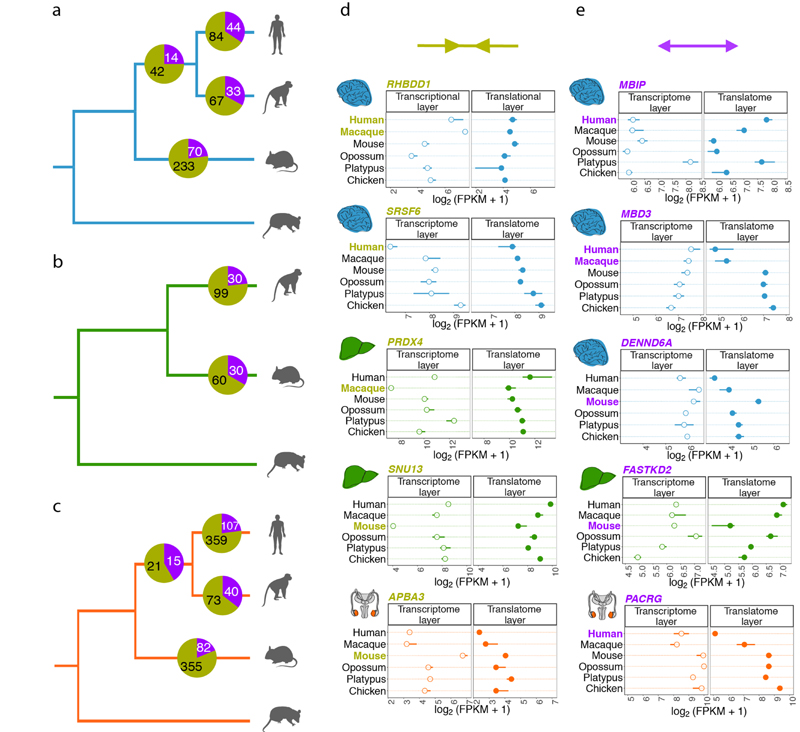
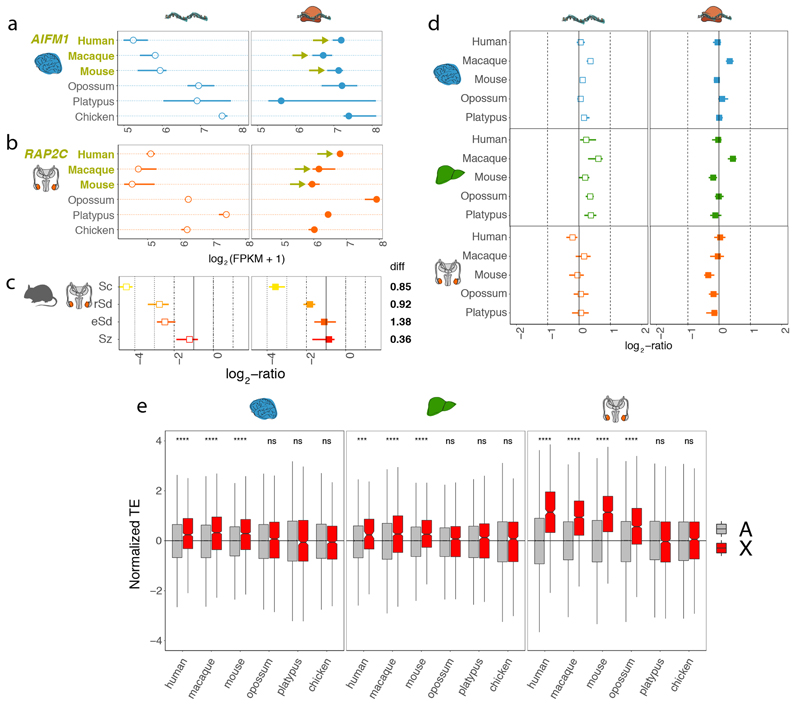
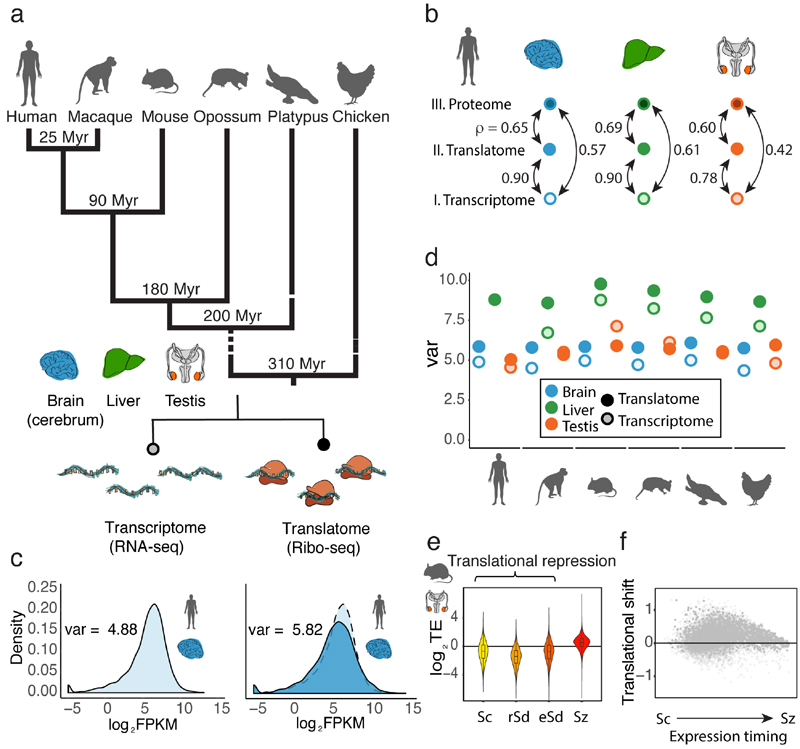
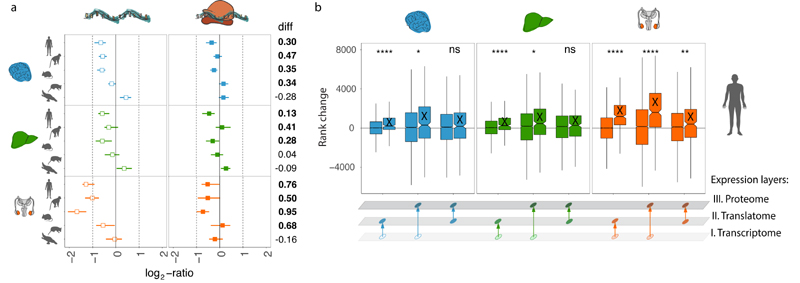
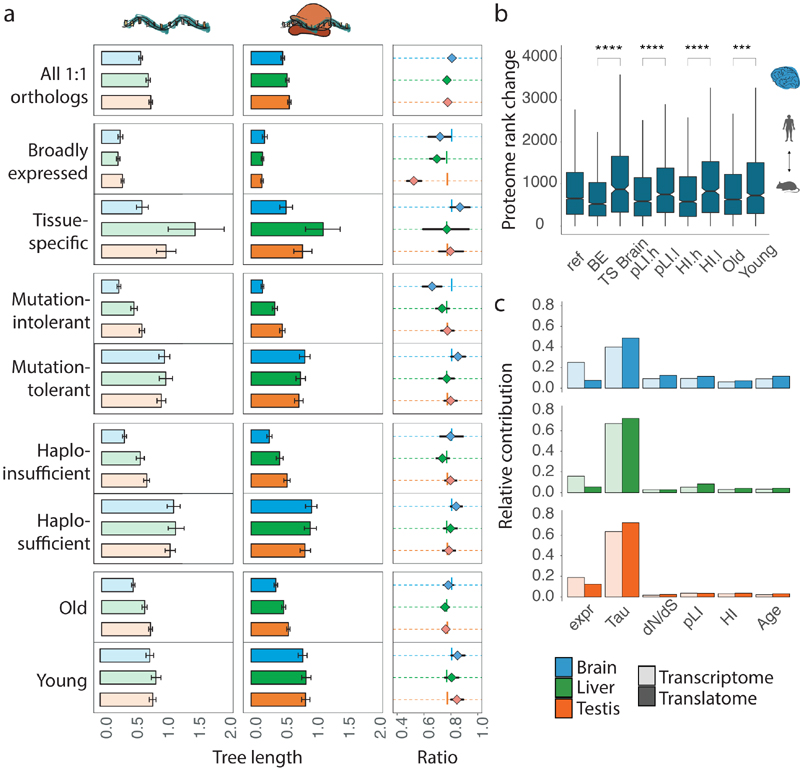
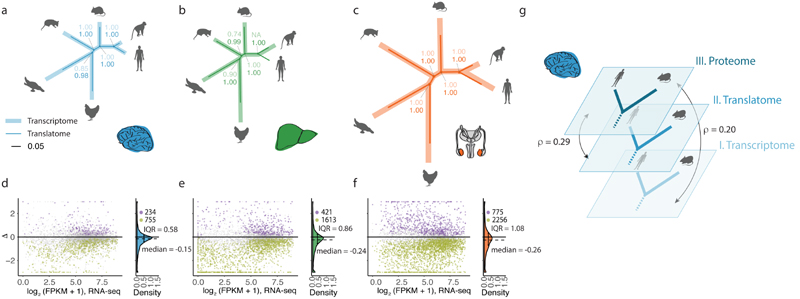
Gene-expression programs define shared and species-specific phenotypes, but their evolution remains largely uncharacterized beyond the transcriptome layer1. Here we report an analysis of the co-evolution of translatomes and transcriptomes using ribosome-profiling and matched RNA-sequencing data for three organs (brain, liver and testis) in five mammals (human, macaque, mouse, opossum and platypus) and a bird (chicken). Our within-species analyses reveal that translational regulation is widespread in the different organs, in particular across the spermatogenic cell types of the testis. The between-species divergence in gene expression is around 20% lower at the translatome layer than at the transcriptome layer owing to extensive buffering between the expression layers, which especially preserved old, essential and housekeeping genes. Translational upregulation specifically counterbalanced global dosage reductions during the evolution of sex chromosomes and the effects of meiotic sex-chromosome inactivation during spermatogenesis. Despite the overall prevalence of buffering, some genes evolved faster at the translatome layer-potentially indicating adaptive changes in expression; testis tissue shows the highest fraction of such genes. Further analyses incorporating mass spectrometry proteomics data establish that the co-evolution of transcriptomes and translatomes is reflected at the proteome layer. Together, our work uncovers co-evolutionary patterns and associated selective forces across the expression layers, and provides a resource for understanding their interplay in mammalian organs.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Necsulea A, Kaessmann H. Evolutionary dynamics of coding and non-coding transcriptomes. Nat Rev Genet. 2014;15:734–748. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
