

Illuminating an Ecological Blackbox: Using High Throughput Sequencing to Characterize the Plant Virome Across Scales
- PMID: 33178159
- PMCID: PMC7596190
- DOI: 10.3389/fmicb.2020.578064
Illuminating an Ecological Blackbox: Using High Throughput Sequencing to Characterize the Plant Virome Across Scales
Abstract
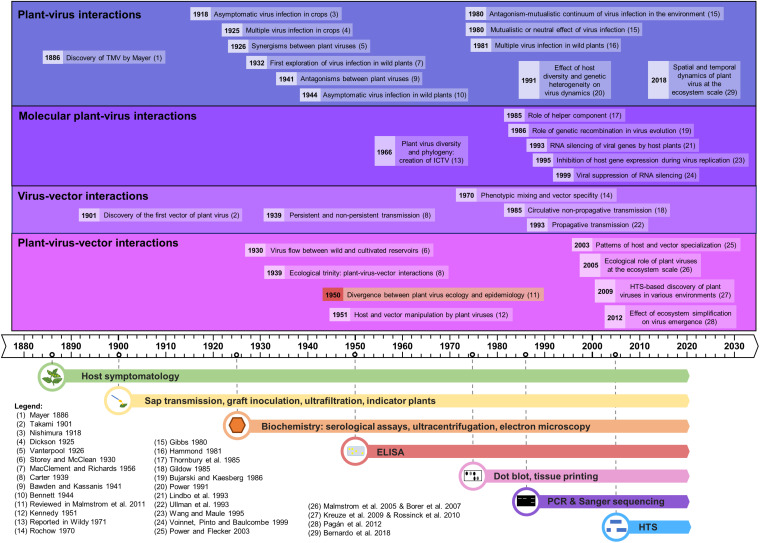
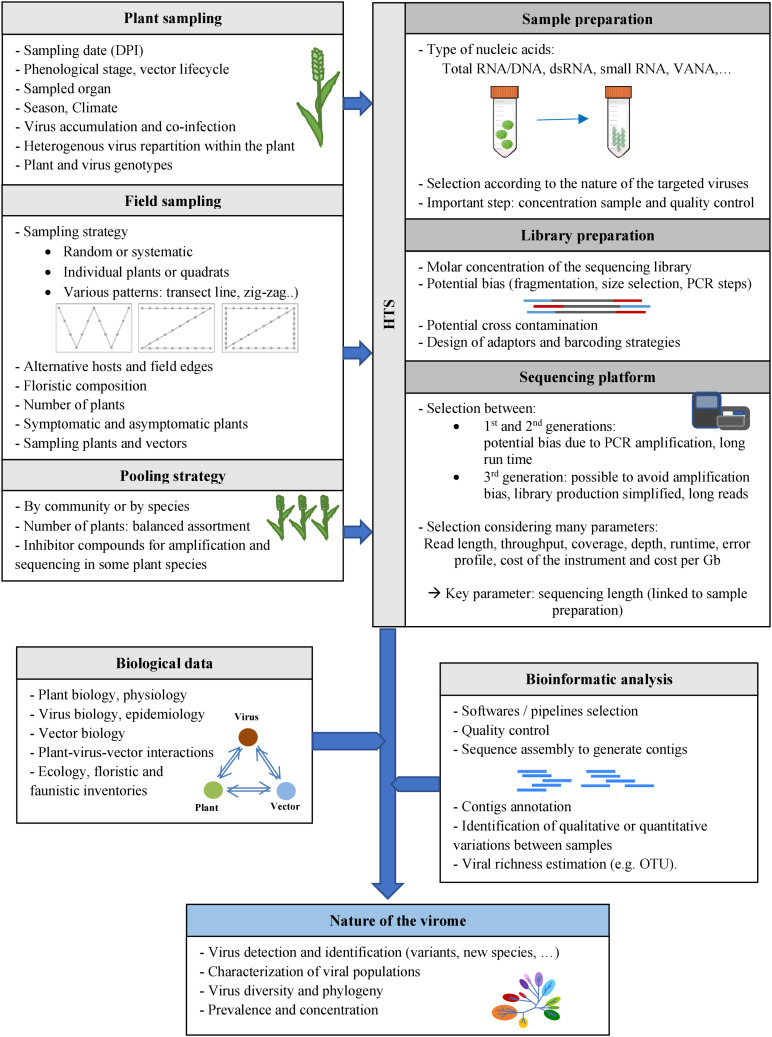
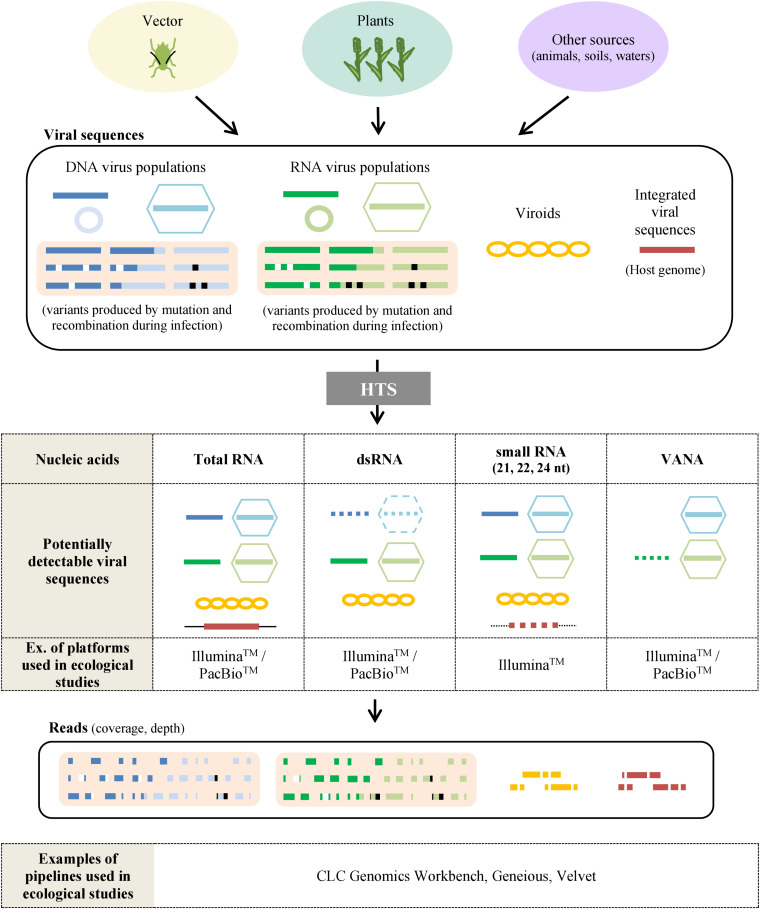
The ecology of plant viruses began to be explored at the end of the 19th century. Since then, major advances have revealed mechanisms of virus-host-vector interactions in various environments. These advances have been accelerated by new technlogies for virus detection and characterization, most recently including high throughput sequencing (HTS). HTS allows investigators, for the first time, to characterize all or nearly all viruses in a sample without a priori information about which viruses might be present. This powerful approach has spurred new investigation of the viral metagenome (virome). The rich virome datasets accumulated illuminate important ecological phenomena such as virus spread among host reservoirs (wild and domestic), effects of ecosystem simplification caused by human activities (and agriculture) on the biodiversity and the emergence of new viruses in crops. To be effective, however, HTS-based virome studies must successfully navigate challenges and pitfalls at each procedural step, from plant sampling to library preparation and bioinformatic analyses. This review summarizes major advances in plant virus ecology associated with technological developments, and then presents important considerations and best practices for HTS use in virome studies.
Keywords: high throughput sequencing; historical advances; opportunities and challenges; plant virome; virus ecology and evolution.
Copyright © 2020 Maclot, Candresse, Filloux, Malmstrom, Roumagnac, van der Vlugt and Massart.
Figures
Similar articles
-
[From boots on the ground to nucleotides in the sequencer: a century of advances in the study of the plant virus ecology].Virologie (Montrouge). 2021 Feb 1;25(1):29-42. doi: 10.1684/vir.2021.0879. Virologie (Montrouge). 2021. PMID: 33650495 Review. French.
-
Long-Term Anthropogenic Management and Associated Loss of Plant Diversity Deeply Impact Virome Richness and Composition of Poaceae Communities.Microbiol Spectr. 2023 Mar 14;11(2):e0485022. doi: 10.1128/spectrum.04850-22. Online ahead of print. Microbiol Spectr. 2023. PMID: 36916941 Free PMC article.
-
Deciphering the virome of Chunkung (Cnidium officinale) showing dwarfism-like symptoms via a high-throughput sequencing analysis.Virol J. 2024 Apr 15;21(1):86. doi: 10.1186/s12985-024-02361-7. Virol J. 2024. PMID: 38622686 Free PMC article.
-
Evolution of selective-sequencing approaches for virus discovery and virome analysis.Virus Res. 2017 Jul 15;239:172-179. doi: 10.1016/j.virusres.2017.06.005. Epub 2017 Jun 3. Virus Res. 2017. PMID: 28583442 Free PMC article. Review.
-
Complexity and Local Specificity of the Virome Associated with Tospovirus-Transmitting Thrips Species.J Virol. 2021 Oct 13;95(21):e0059721. doi: 10.1128/JVI.00597-21. Epub 2021 Jul 7. J Virol. 2021. PMID: 34232724 Free PMC article.
Cited by
-
Identification and complete genome sequencing of a divergent olive virus T isolate and an olive leaf yellowing-associated virus isolate naturally infecting olive trees in Greece.Virus Genes. 2022 Dec;58(6):560-569. doi: 10.1007/s11262-022-01934-4. Epub 2022 Sep 24. Virus Genes. 2022. PMID: 36152231 Free PMC article.
-
Reproducibility and Sensitivity of High-Throughput Sequencing (HTS)-Based Detection of Citrus Tristeza Virus and Three Citrus Viroids.Plants (Basel). 2022 Jul 26;11(15):1939. doi: 10.3390/plants11151939. Plants (Basel). 2022. PMID: 35893644 Free PMC article.
-
The Sisal Virome: Uncovering the Viral Diversity of Agave Varieties Reveals New and Organ-Specific Viruses.Microorganisms. 2021 Aug 10;9(8):1704. doi: 10.3390/microorganisms9081704. Microorganisms. 2021. PMID: 34442783 Free PMC article.
-
Expanding the environmental virome: Infection profile in a native rainforest tree species.Front Microbiol. 2022 Aug 4;13:874319. doi: 10.3389/fmicb.2022.874319. eCollection 2022. Front Microbiol. 2022. PMID: 35992690 Free PMC article.
-
Tobacco Mild Green Mosaic Virus (TMGMV) Isolates from Different Plant Families Show No Evidence of Differential Adaptation to Their Host of Origin.Viruses. 2023 Dec 5;15(12):2384. doi: 10.3390/v15122384. Viruses. 2023. PMID: 38140625 Free PMC article.
References
-
- Abarshi M. M., Mohammed I. U., Wasswa P., Hillocks R. J., Holt J., Legg J. P., et al. (2010). Optimization of diagnostic RT-PCR protocols and sampling procedures for the reliable and cost-effective detection of Cassava brown streak virus. J. Virol. Methods 163 353–359. 10.1016/j.jviromet.2009.10.023 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources