

Clinical, Genetic, and Prognostic Features of Adrenocortical Tumors in Children: A 10-Year Single-Center Experience
- PMID: 33178583
- PMCID: PMC7593337
- DOI: 10.3389/fonc.2020.554388
Clinical, Genetic, and Prognostic Features of Adrenocortical Tumors in Children: A 10-Year Single-Center Experience
Abstract
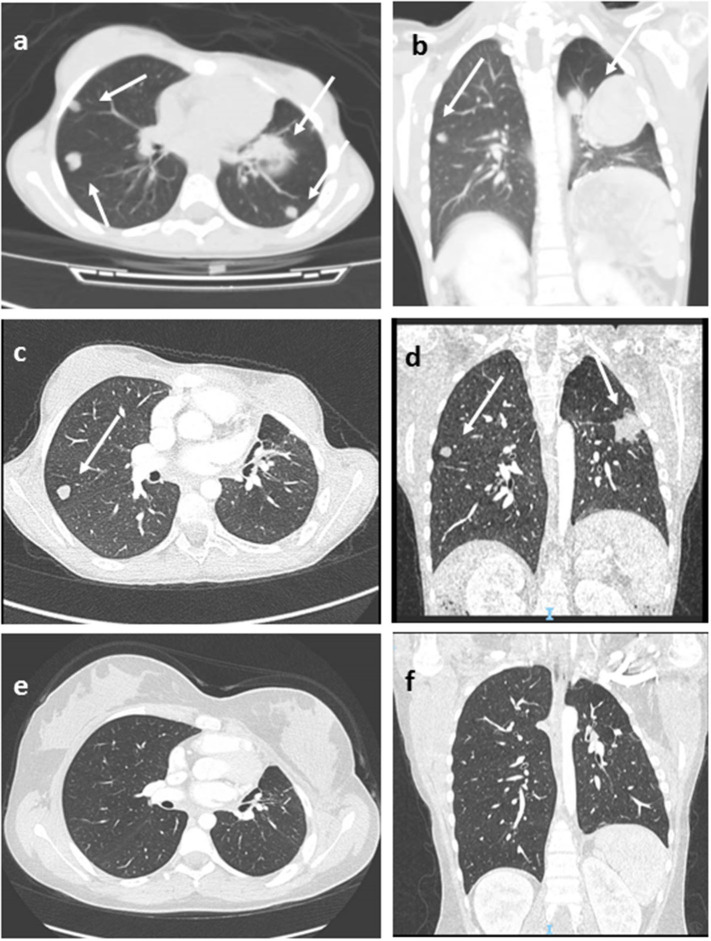
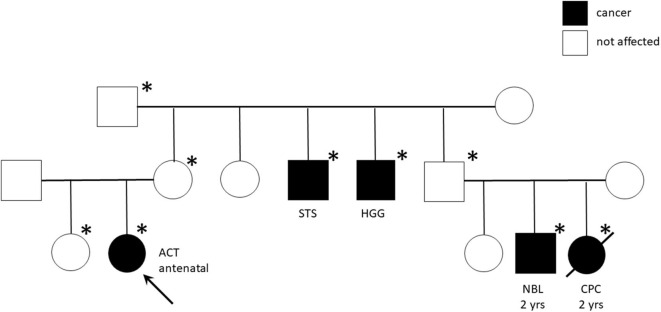
Background and Aims: Pediatric adrenocortical tumors (ACTs) are very rare endocrine neoplasms in childhood. In this study, we performed a retrospective analysis of children with ACT treated at our institution by examining clinical and genetic disease features, treatment strategies, and outcomes. Methods: We retrospectively analyzed a cohort of 13 children treated at the Bambino Gesù Children's Hospital from November 2010 to March 2020. Results: The median age at diagnosis was 17 months (range = 0-82 months). The female: male ratio was 3.3/1. Mixed symptomatology (>1 hormone abnormality) was the most common presentation (46.1%). In three cases, the tumor was detected during prenatal or perinatal echographic screening. All patients presented with localized disease at diagnosis and underwent total adrenalectomy. Six patients were identified as having malignancies according to the Wieneke scoring system, five benign, and two undetermined. Seven patients underwent mitotane adjuvant therapy for 12 months. There was metastatic disease in three patients, with no correlation with age or Wieneke score. The most common sites of metastases were the liver and lungs. Metastatic patients were treated with surgery (n = 2), mitotane (n = 1), chemotherapy (n = 2) associated with anti-EGFR (n = 1), or immunotherapy with anti-PD1 (pembrolizumab) (n = 1); two patients achieved complete disease remission. Overall 2- and 5-year survival rates were 100%, with a median follow-up of 5 years (range = 2-9.5 years). Two- and 5-year disease free survival was 76.9 and 84.6%, respectively (95% confidence interval = -66.78-114.76 months). All patients are alive, 12 without disease, and one with stable disease. Genetic analyses showed TP53 germline mutations in six of eight patients analyzed (five inherited, one de novo). One patient had Beckwith-Wiedemann syndrome, with mosaic paternal uniparental disomy of chromosome 11, in both neoplastic and healthy adrenal tissue. Conclusion: We report the cases of 13 patients treated for ACT, including 12 aged <4 years at diagnosis, with a relative short time from symptoms onset. Our cohort experienced an excellent prognosis. TP53 mutation was found in 75% of tested patients (6/8) confirming the need to perform genetic tests and familial counseling in this disease.
Keywords: Beckwith–Wiedeman syndrome; Li-Fraumeni Syndrome; adrenocortical tumors; children; immunotherapy; mitotane; prognosis; targeted therapies.
Copyright © 2020 Miele, Di Giannatale, Crocoli, Cozza, Serra, Castellano, Cacchione, Cefalo, Alaggio and De Pasquale.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous