

Primary hyperammonaemia: Current diagnostic and therapeutic strategies
- PMID: 33179600
- PMCID: PMC8518097
- DOI: 10.34763/jmotherandchild.20202402si.2015.000006
Primary hyperammonaemia: Current diagnostic and therapeutic strategies
Abstract
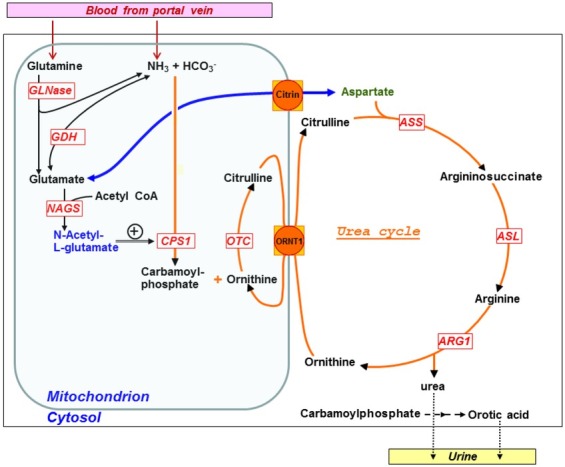
Primary hyperammonaemia is a term to describe an elevation of ammonia in blood or plasma due to a defect within the urea cycle, which is the pathway responsible for ammonia detoxification and arginine biosynthesis. Urea cycle disorders (UCDs) are rare diseases caused by genetic defects affecting any of the six enzymes or two transporters that are directly involved in the urea cycle function.The clinical situation is variable and largely depends on the time of onset. Newborns who are often affected by hyper-ammonaemic encephalopathy carry a potential risk of severe brain damage, which may lead to death. Outside the neonatal period, symptoms are very unspecific but most often neurological (with wide variability), psychiatric and/or gastrointestinal. Early identification of patients is extremely important to start effective treatment modalities immediately. The acute management includes detoxification of ammonia, which often requires extracorporeal means such as haemodialysis, and the use of intravenous drugs that work as nitrogen scavengers. Long-term management of patients with UCDs consists of a low-protein diet, which needs to be balanced and supplemented to avoid deficiencies of essential amino acids, trace elements or vitamins and the use of nitrogen scavengers.The reader will find here a brief overview describing the most relevant aspects of the clinical management of UCDs in an attempt to raise awareness for this important group of rare diseases.
Keywords: encephalopathy; haemodialysis; hyperammonaemia; low-protein diet; nitrogen metabolism; nitrogen scavengers; urea cycle.
Conflict of interest statement
None.
Figures
References
-
- Häberle J, Rubio V. Saudubray J-M, Baumgartner MR, Walter JH. Inborn metabolic diseases: diagnosis and treatment. 6th ed. Berlin, Heidelberg: Springer; 2016. Disorders of the urea cycle and related enzymes; pp. 295–308.pp. 658
-
- Ah Mew N, Simpson KL, Gropman AL, Lanpher BC, Chapman KA, Summar ML. Urea cycle disorders overview. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A. Genetic counseling, editors. GeneReviews® [Internet] Seattle, WA: University of Washington;; 2003. 1993–2020. Apr 29 [updated 2017 Jun 22] ISSN 2372-0697.
-
- Tuchman M. The clinical, biochemical, and molecular spectrum of ornithine transcarbamylase deficiency. J Lab Clin Med. 1992;120(6):836–50. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical