

An autonomous, but INSIG-modulated, role for the sterol sensing domain in mallostery-regulated ERAD of yeast HMG-CoA reductase
- PMID: 33184059
- PMCID: PMC7948459
- DOI: 10.1074/jbc.RA120.015910
An autonomous, but INSIG-modulated, role for the sterol sensing domain in mallostery-regulated ERAD of yeast HMG-CoA reductase
Abstract
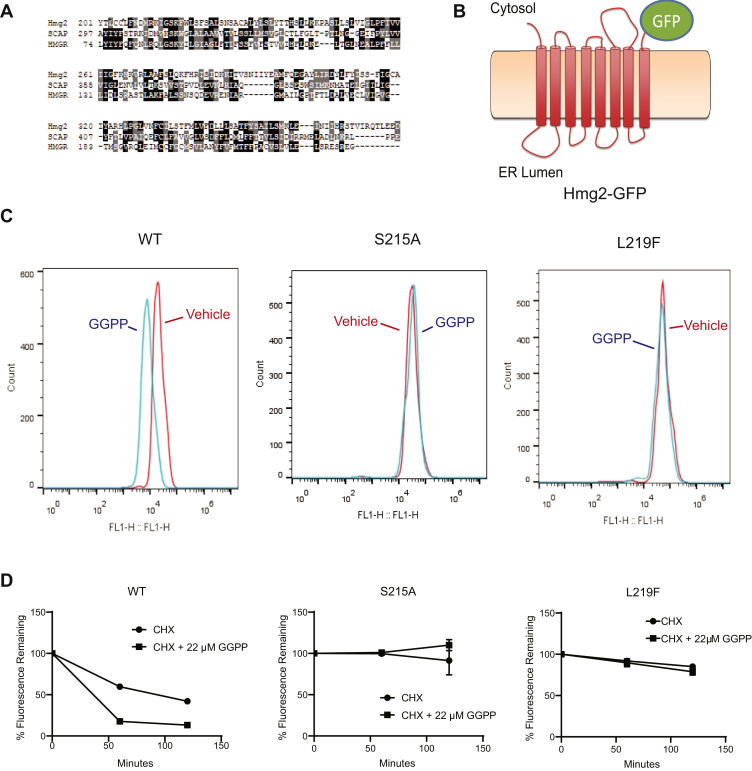
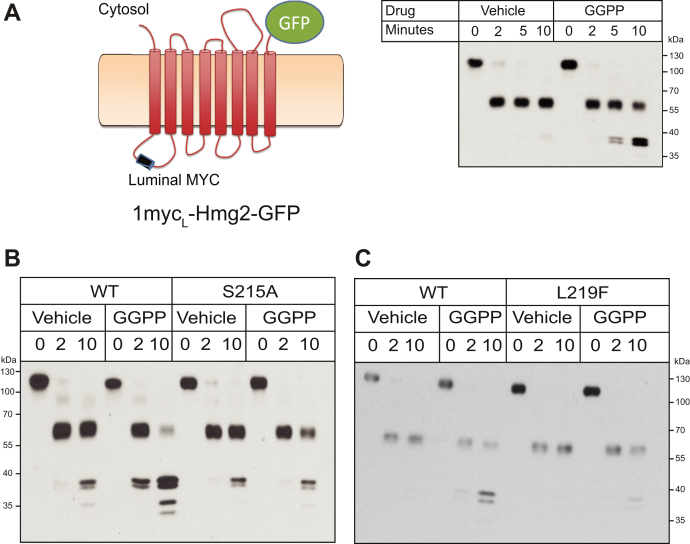
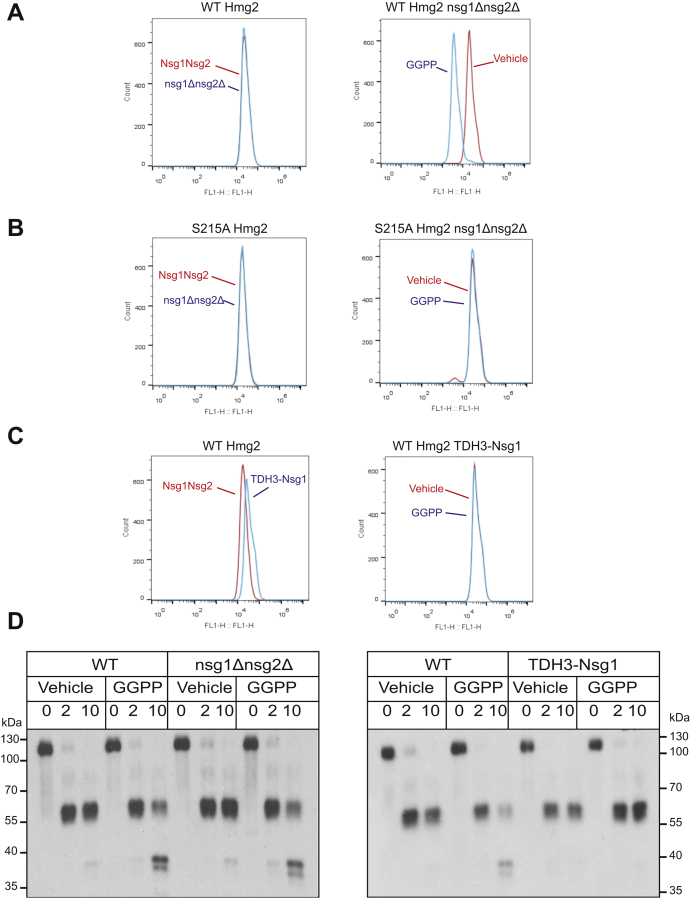
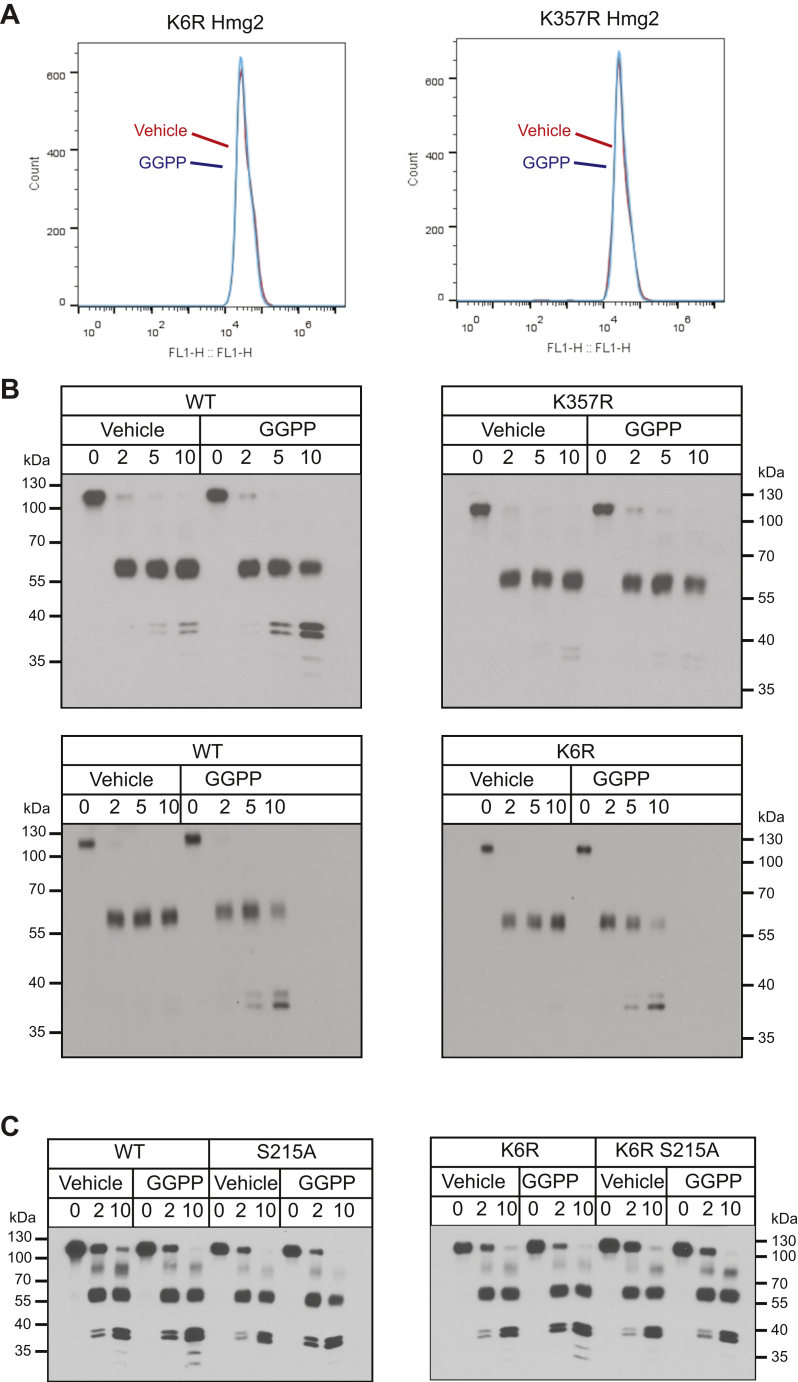
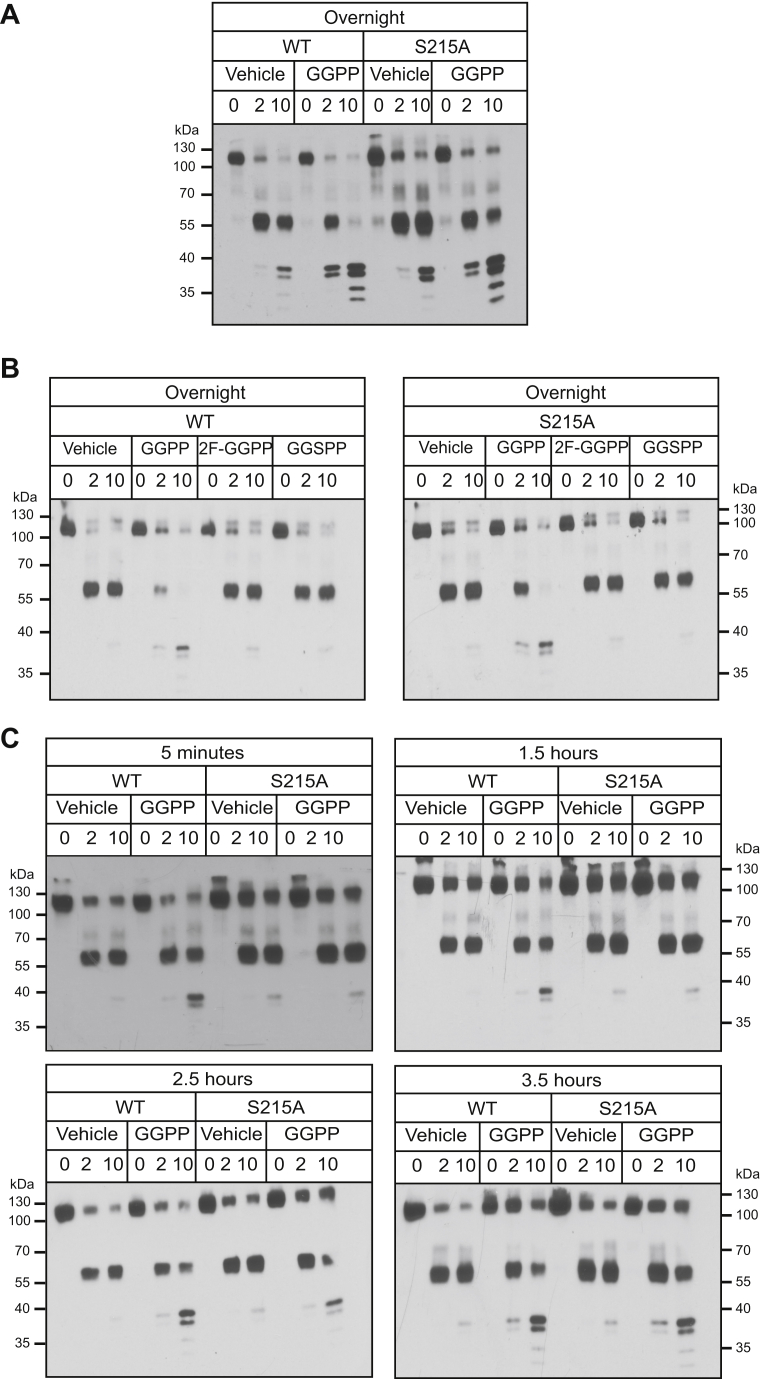
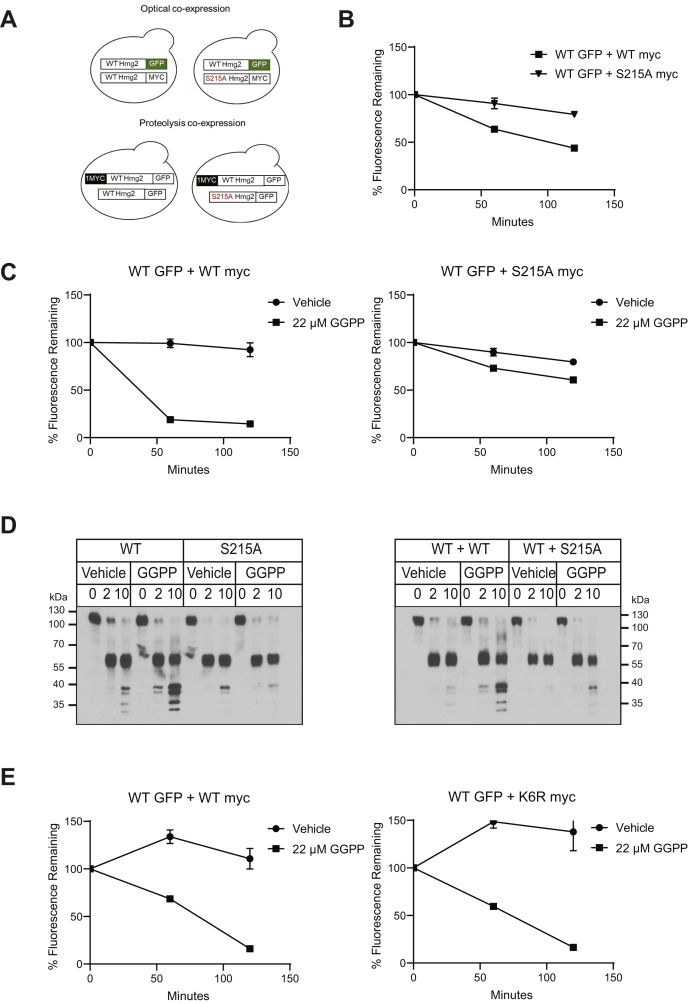
HMG-CoA reductase (HMGR) undergoes feedback-regulated degradation as part of sterol pathway control. Degradation of the yeast HMGR isozyme Hmg2 is controlled by the sterol pathway intermediate GGPP, which causes misfolding of Hmg2, leading to degradation by the HRD pathway; we call this process mallostery. We evaluated the role of the Hmg2 sterol sensing domain (SSD) in mallostery, as well as the involvement of the highly conserved INSIG proteins. We show that the Hmg2 SSD is critical for regulated degradation of Hmg2 and required for mallosteric misfolding of GGPP as studied by in vitro limited proteolysis. The Hmg2 SSD functions independently of conserved yeast INSIG proteins, but its function was modulated by INSIG, thus imposing a second layer of control on Hmg2 regulation. Mutant analyses indicated that SSD-mediated mallostery occurred prior to and independent of HRD-dependent ubiquitination. GGPP-dependent misfolding was still extant but occurred at a much slower rate in the absence of a functional SSD, indicating that the SSD facilitates a physiologically useful rate of GGPP response and implying that the SSD is not a binding site for GGPP. Nonfunctional SSD mutants allowed us to test the importance of Hmg2 quaternary structure in mallostery: a nonresponsive Hmg2 SSD mutant strongly suppressed regulation of a coexpressed, normal Hmg2. Finally, we have found that GGPP-regulated misfolding occurred in detergent-solubilized Hmg2, a feature that will allow next-level analysis of the mechanism of this novel tactic of ligand-regulated misfolding.
Keywords: ER quality control; HMG-CoA reductase; HRD pathway; cholesterol regulation; endoplasmic-reticulum-associated protein degradation (ERAD); mallostery; protein misfolding; sterol sensing domain (SSD); ubiquitin; ubiquitin-proteasome system.
Copyright © 2021. Published by Elsevier Inc.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures

References
-
- Mehnert M., Sommer T., Jarosch E. ERAD ubiquitin ligases. BioEssays. 2010;32:905–913. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
