

Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer
- PMID: 33184106
- PMCID: PMC7854521
- DOI: 10.1158/0008-5472.CAN-20-1200
Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer
Abstract
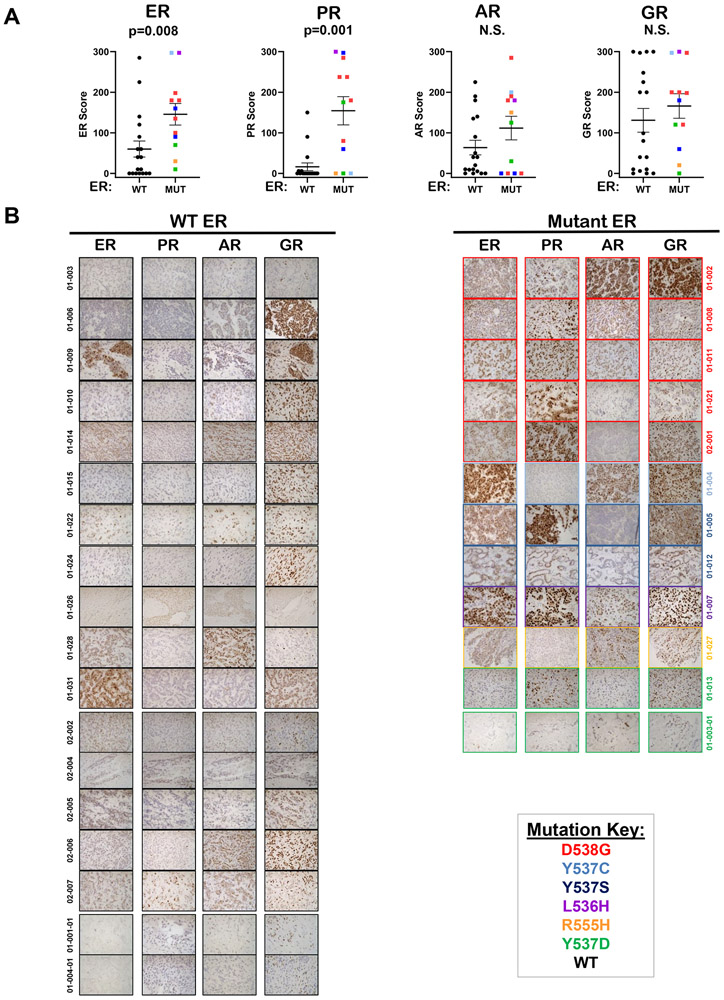
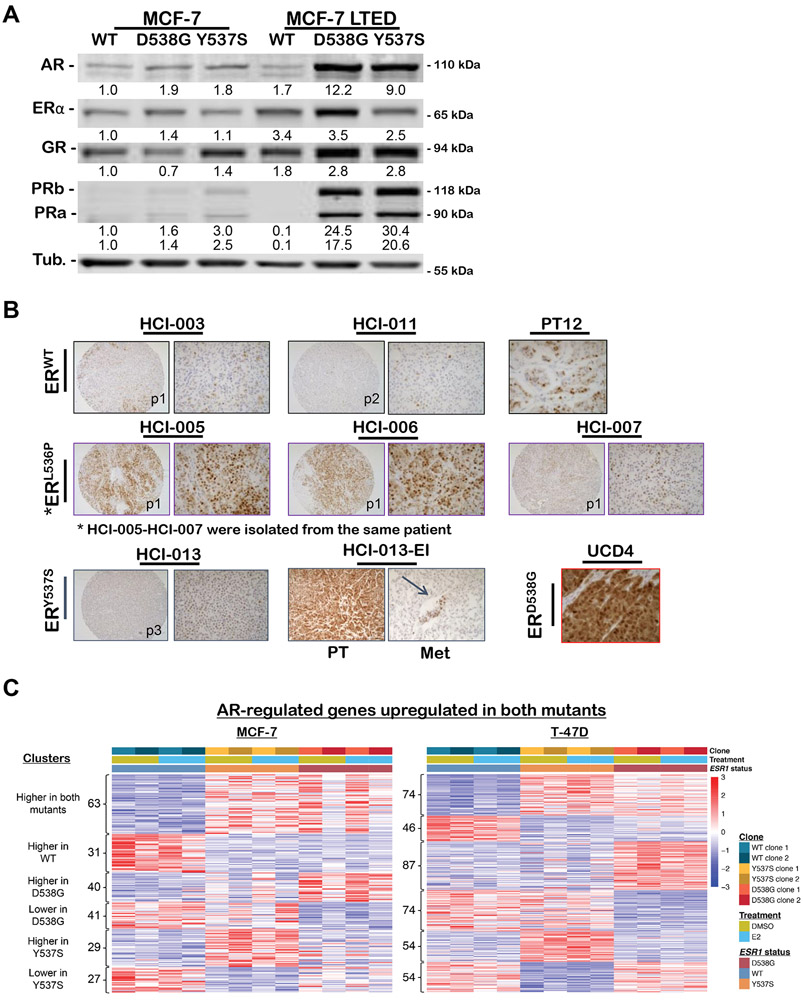
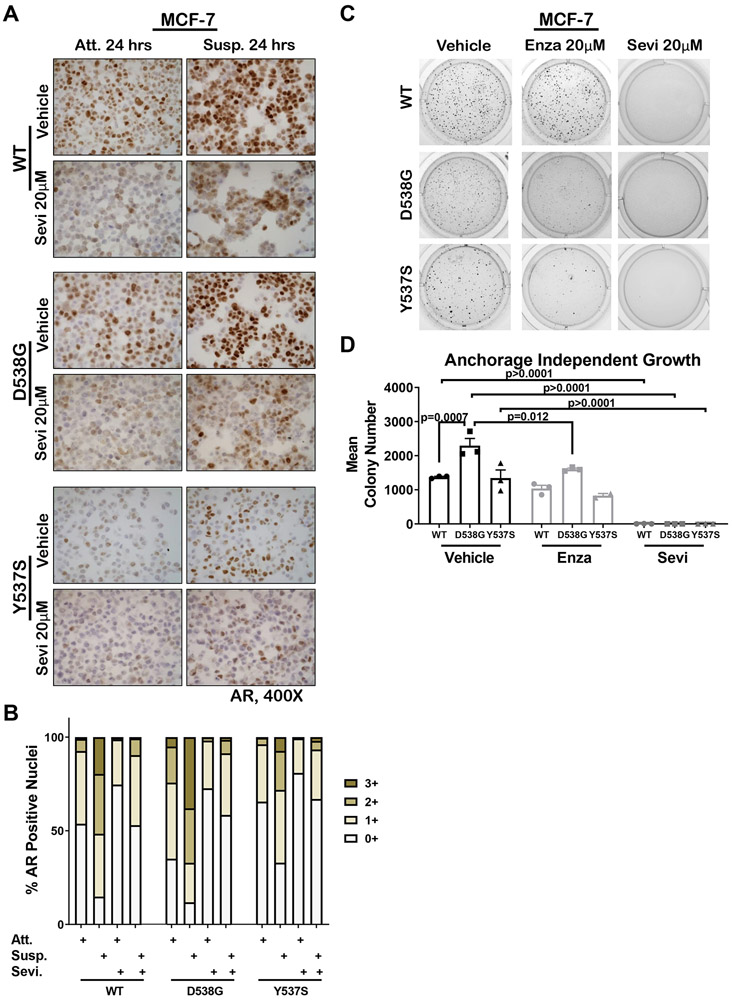
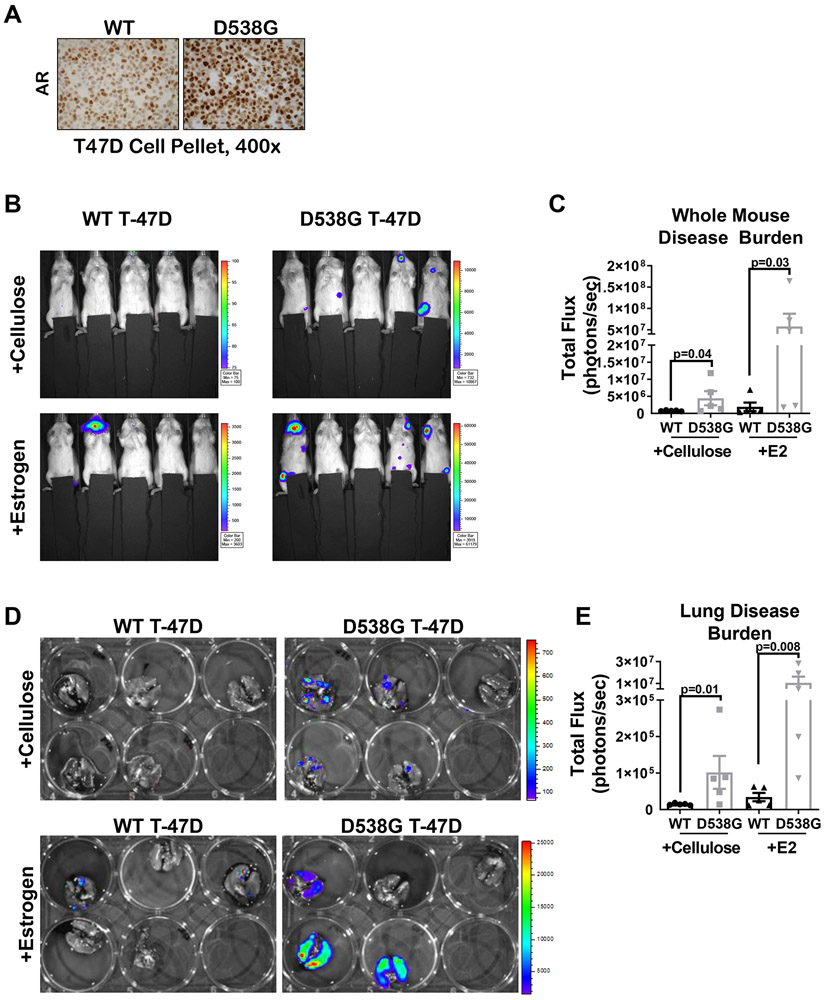
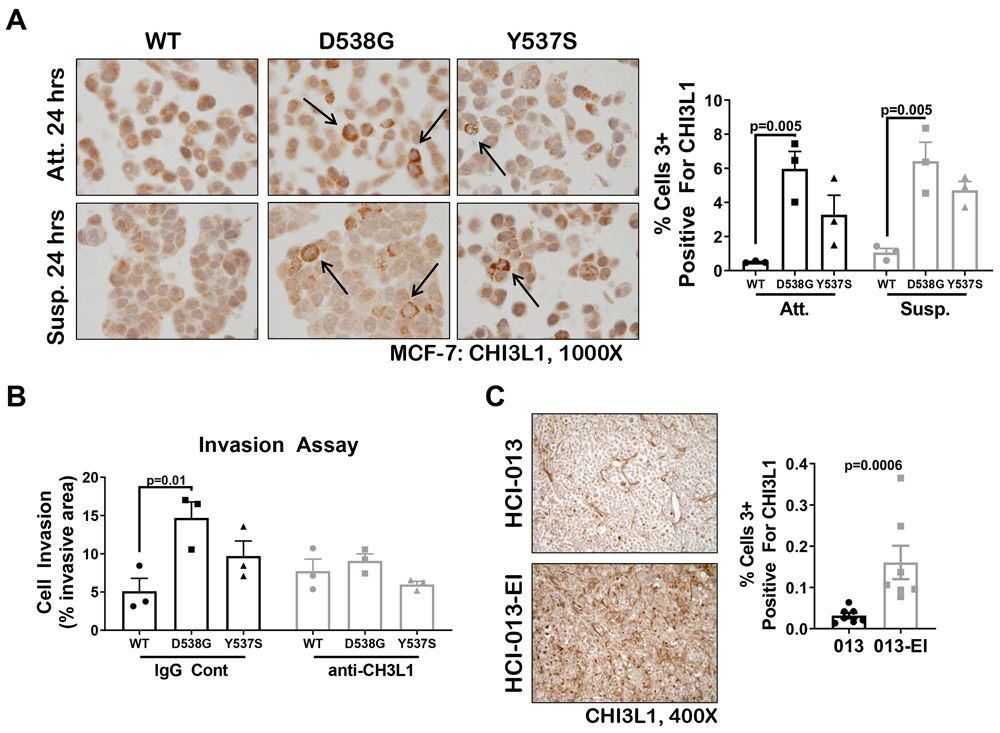
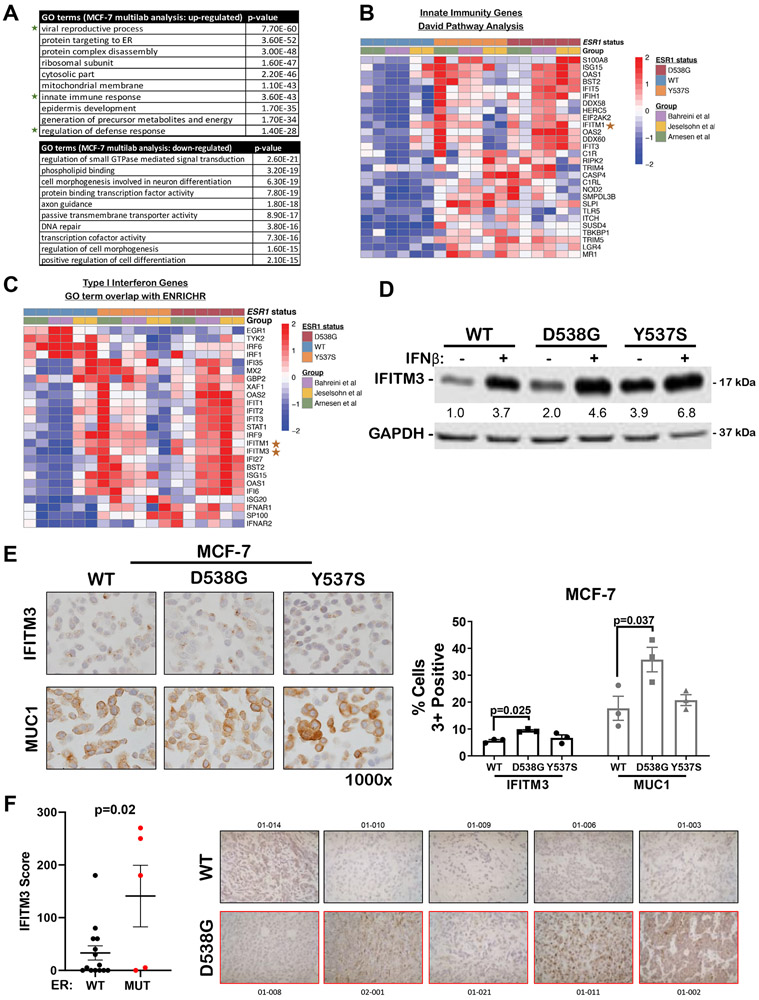
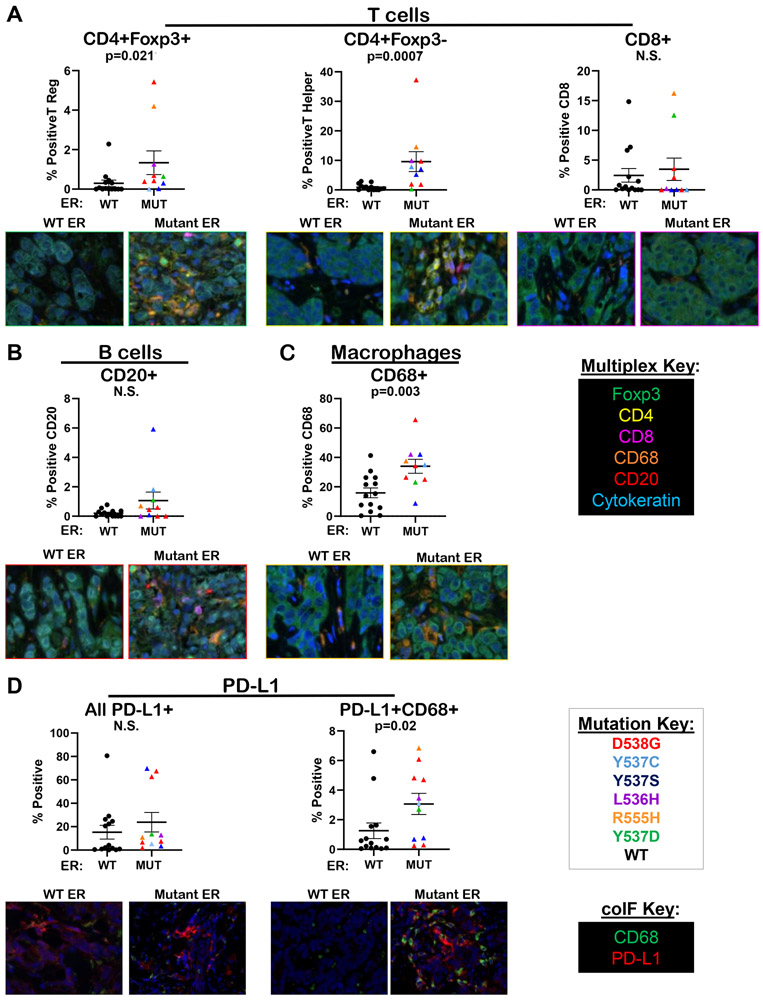
Mutations in ESR1 that confer constitutive estrogen receptor alpha (ER) activity in the absence of ligand are acquired by ≥40% of metastatic breast cancers (MBC) resistant to adjuvant aromatase inhibitor (AI) therapy. To identify targetable vulnerabilities in MBC, we examined steroid hormone receptors and tumor-infiltrating immune cells in metastatic lesions with or without ER mutations. ER and progesterone receptor (PR) were significantly lower in metastases with wild-type (WT) ER compared with those with mutant ER, suggesting that metastases that evade AI therapy by mechanism(s) other than acquiring ER mutations lose dependency on ER and PR. Metastases with mutant ER had significantly higher T regulatory and Th cells, total macrophages, and programmed death ligand-1 (PD-L1)-positive immune-suppressive macrophages than those with WT ER. Breast cancer cells with CRISPR-Cas9-edited ER (D538G, Y537S, or WT) and patient-derived xenografts harboring mutant or WT ER revealed genes and proteins elevated in mutant ER cells, including androgen receptor (AR), chitinase-3-like protein 1 (CHI3L1), and IFN-stimulated genes (ISG). Targeting these proteins blunted the selective advantage of ER-mutant tumor cells to survive estrogen deprivation, anchorage independence, and invasion. Thus, patients with mutant ER MBC might respond to standard-of-care fulvestrant or other selective ER degraders when combined with AR or CHI3L1 inhibition, perhaps with the addition of immunotherapy. SIGNIFICANCE: Targetable alterations in MBC, including AR, CHI3L1, and ISG, arise following estrogen-deprivation, and ER-mutant metastases may respond to immunotherapies due to elevated PD-L1+ macrophages.See related article by Arnesen et al., p. 539.
©2020 American Association for Cancer Research.
Conflict of interest statement
Figures
Comment on
-
Estrogen Receptor Alpha Mutations in Breast Cancer Cells Cause Gene Expression Changes through Constant Activity and Secondary Effects.Cancer Res. 2021 Feb 1;81(3):539-551. doi: 10.1158/0008-5472.CAN-20-1171. Epub 2020 Nov 12. Cancer Res. 2021. PMID: 33184109 Free PMC article.
Similar articles
-
Longitudinal Molecular Imaging of Progesterone Receptor Reveals Early Differential Response to Endocrine Therapy in Breast Cancer with an Activating ESR1 Mutation.J Nucl Med. 2021 Apr;62(4):500-506. doi: 10.2967/jnumed.120.249508. Epub 2020 Aug 28. J Nucl Med. 2021. PMID: 32859700 Free PMC article.
-
The prevalence of estrogen receptor-1 mutation in advanced breast cancer: The estrogen receptor one study (EROS1).Cancer Treat Res Commun. 2019;19:100123. doi: 10.1016/j.ctarc.2019.100123. Epub 2019 Feb 21. Cancer Treat Res Commun. 2019. PMID: 30826563
-
Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models.Breast Cancer Res. 2017 May 23;19(1):60. doi: 10.1186/s13058-017-0851-4. Breast Cancer Res. 2017. PMID: 28535794 Free PMC article.
-
ESR1 mutations and therapeutic resistance in metastatic breast cancer: progress and remaining challenges.Br J Cancer. 2022 Feb;126(2):174-186. doi: 10.1038/s41416-021-01564-x. Epub 2021 Oct 7. Br J Cancer. 2022. PMID: 34621045 Free PMC article. Review.
-
ESR1 mutations in breast cancer.Cancer. 2019 Nov 1;125(21):3714-3728. doi: 10.1002/cncr.32345. Epub 2019 Jul 18. Cancer. 2019. PMID: 31318440 Free PMC article. Review.
Cited by
-
VIVA1: a more invasive subclone of MDA-MB-134VI invasive lobular carcinoma cells with increased metastatic potential in xenograft models.Br J Cancer. 2022 Jul;127(1):56-68. doi: 10.1038/s41416-022-01778-7. Epub 2022 Mar 22. Br J Cancer. 2022. PMID: 35318435 Free PMC article.
-
Clinical and immune responses to neoadjuvant fulvestrant with or without enzalutamide in ER+/Her2- breast cancer.NPJ Breast Cancer. 2024 Oct 6;10(1):88. doi: 10.1038/s41523-024-00697-5. NPJ Breast Cancer. 2024. PMID: 39368973 Free PMC article.
-
The Great Immune Escape: Understanding the Divergent Immune Response in Breast Cancer Subtypes.Cancer Discov. 2023 Jan 9;13(1):23-40. doi: 10.1158/2159-8290.CD-22-0475. Cancer Discov. 2023. PMID: 36620880 Free PMC article. Review.
-
Reversing an Oncogenic Epithelial-to-Mesenchymal Transition Program in Breast Cancer Reveals Actionable Immune Suppressive Pathways.Pharmaceuticals (Basel). 2021 Nov 2;14(11):1122. doi: 10.3390/ph14111122. Pharmaceuticals (Basel). 2021. PMID: 34832904 Free PMC article. Review.
-
Phase II trial of fulvestrant plus enzalutamide in ER+/HER2- advanced breast cancer.NPJ Breast Cancer. 2023 May 20;9(1):41. doi: 10.1038/s41523-023-00544-z. NPJ Breast Cancer. 2023. PMID: 37210417 Free PMC article.
References
-
- Jeselsohn R, De Angelis C, Brown M, Schiff R. The Evolving Role of the Estrogen Receptor Mutations in Endocrine Therapy-Resistant Breast Cancer. Curr Oncol Rep 2017;19:35. - PubMed
-
- Karnik PS, Kulkarni S, Liu XP, Budd GT, Bukowski RM. Estrogen receptor mutations in tamoxifen-resistant breast cancer. Cancer Res 1994;54:349–53 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
