

Replication Gaps Underlie BRCA Deficiency and Therapy Response
- PMID: 33184108
- PMCID: PMC8026497
- DOI: 10.1158/0008-5472.CAN-20-1602
Replication Gaps Underlie BRCA Deficiency and Therapy Response
Abstract
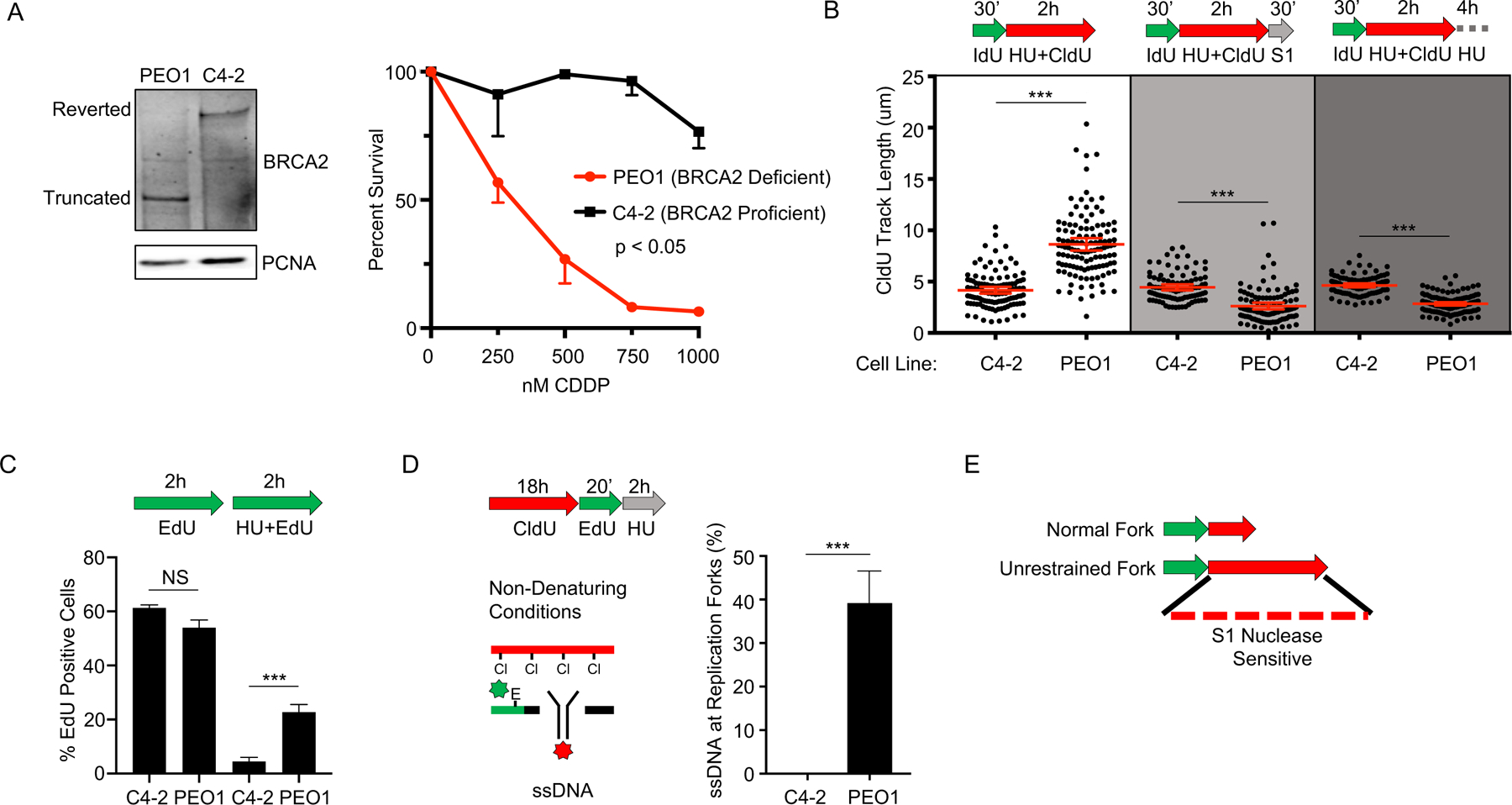
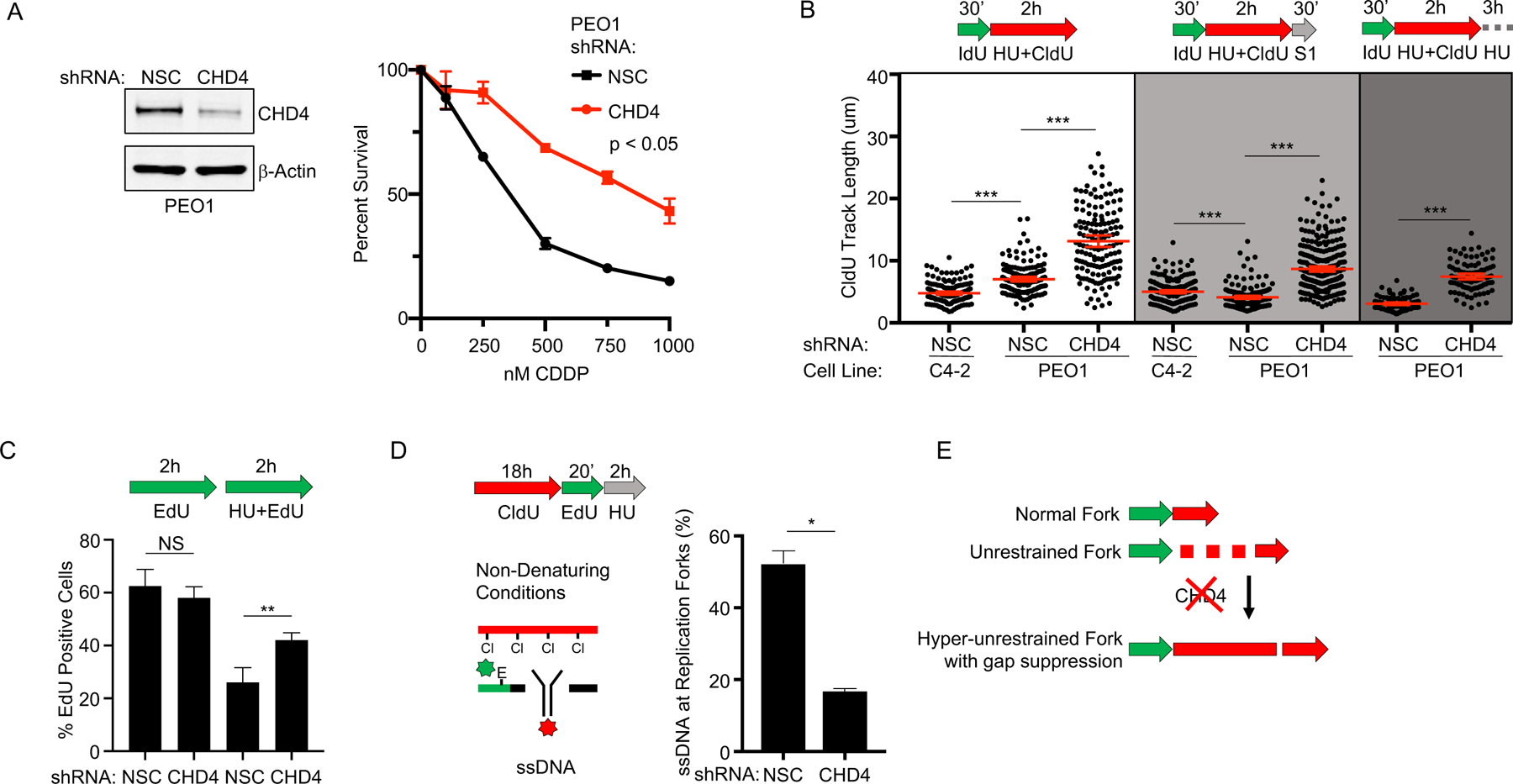
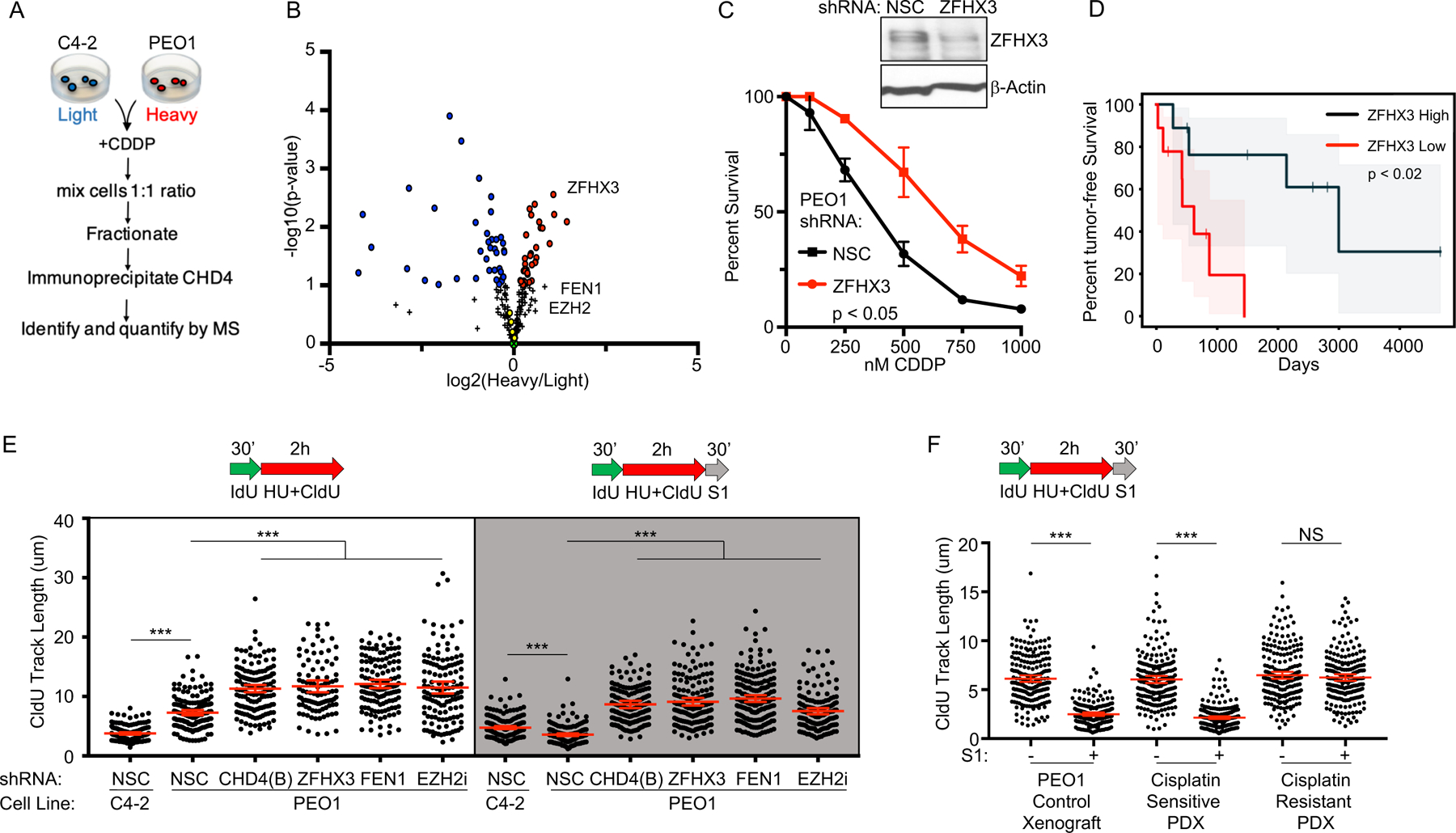
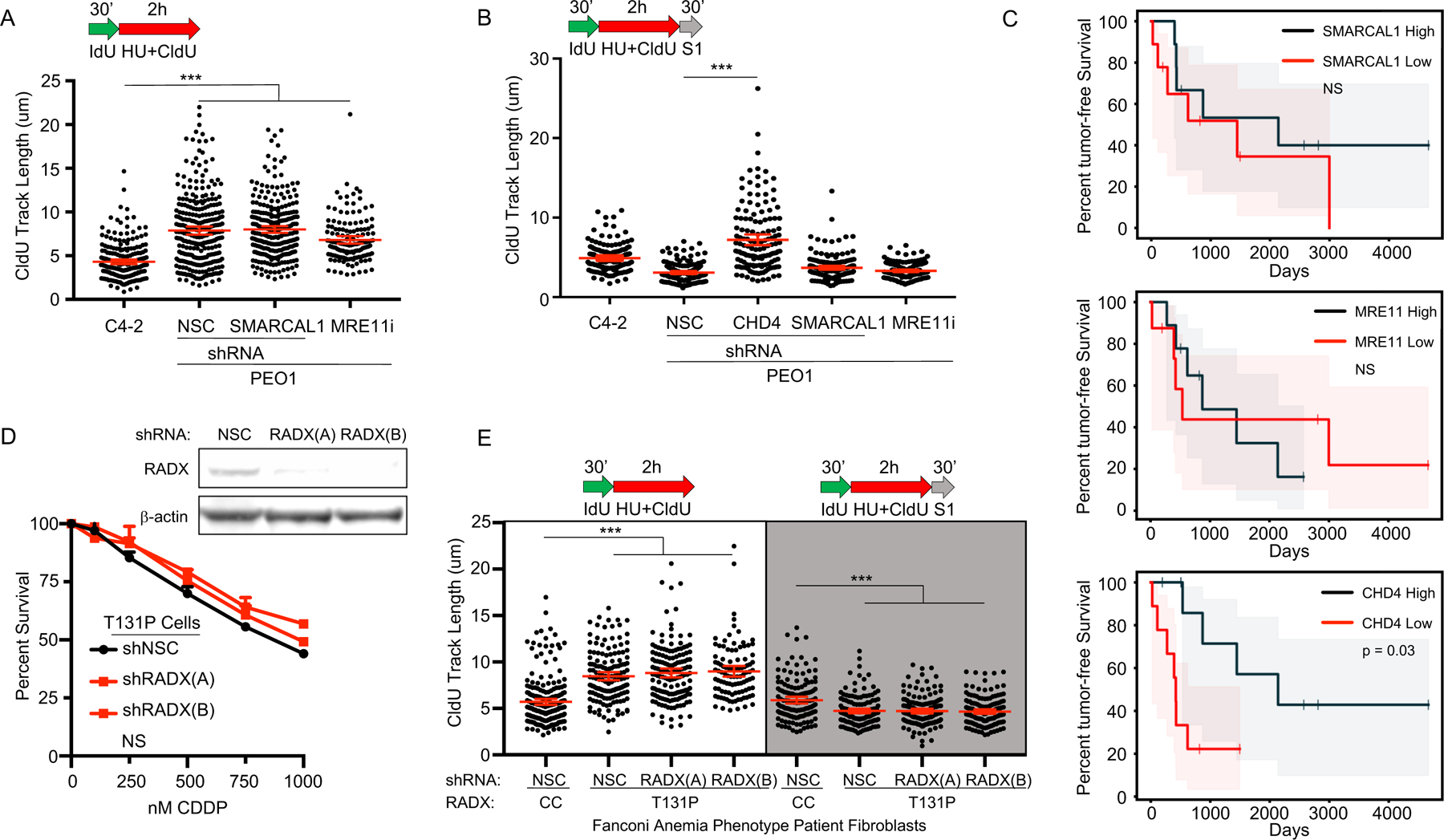
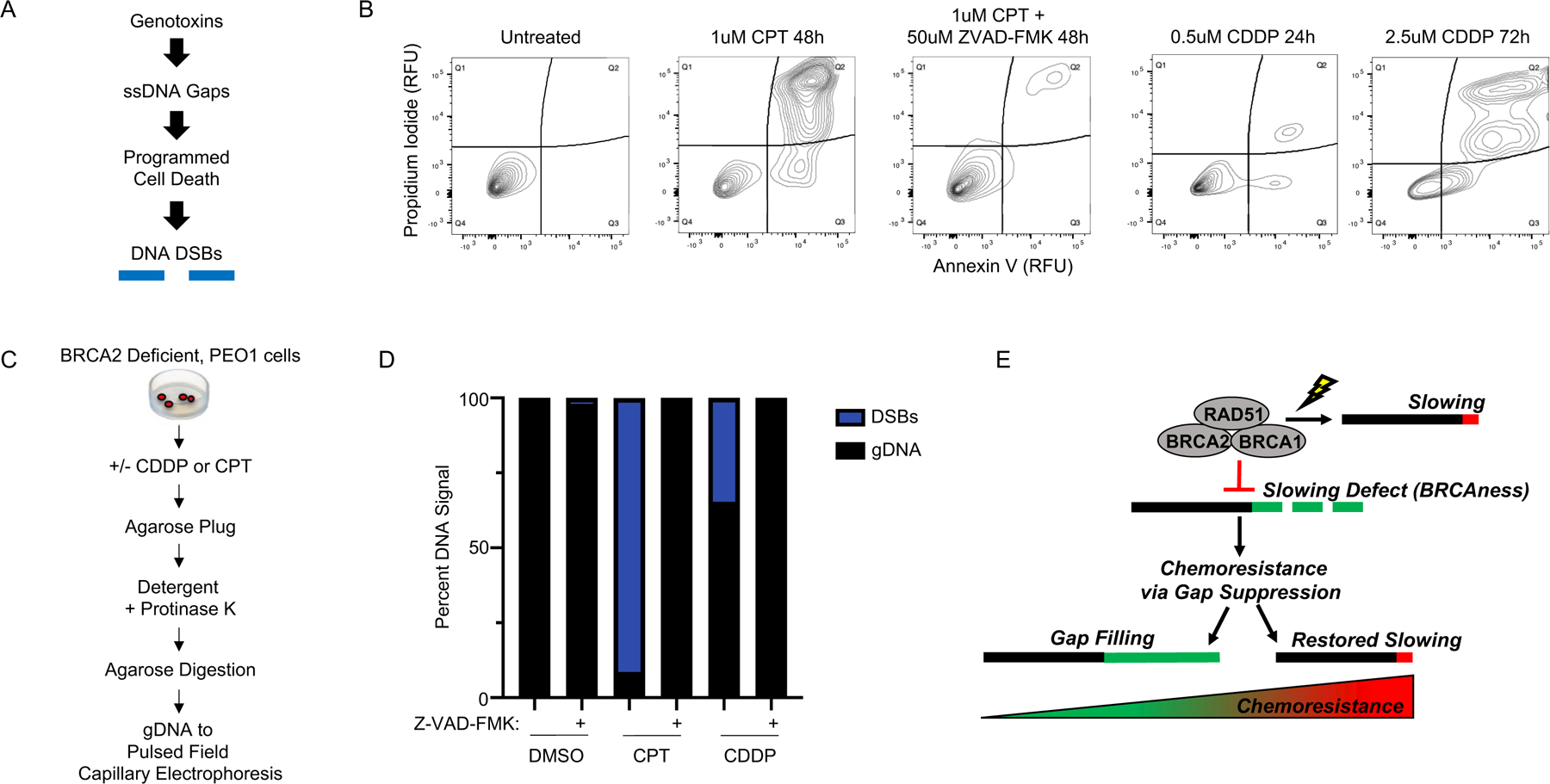
Defects in DNA repair and the protection of stalled DNA replication forks are thought to underlie the chemosensitivity of tumors deficient in the hereditary breast cancer genes BRCA1 and BRCA2 (BRCA). Challenging this assumption are recent findings that indicate chemotherapies, such as cisplatin used to treat BRCA-deficient tumors, do not initially cause DNA double-strand breaks (DSB). Here, we show that ssDNA replication gaps underlie the hypersensitivity of BRCA-deficient cancer and that defects in homologous recombination (HR) or fork protection (FP) do not. In BRCA-deficient cells, ssDNA gaps developed because replication was not effectively restrained in response to stress. Gap suppression by either restoration of fork restraint or gap filling conferred therapy resistance in tissue culture and BRCA patient tumors. In contrast, restored FP and HR could be uncoupled from therapy resistance when gaps were present. Moreover, DSBs were not detected after therapy when apoptosis was inhibited, supporting a framework in which DSBs are not directly induced by genotoxic agents, but rather are induced from cell death nucleases and are not fundamental to the mechanism of action of genotoxic agents. Together, these data indicate that ssDNA replication gaps underlie the BRCA cancer phenotype, "BRCAness," and we propose they are fundamental to the mechanism of action of genotoxic chemotherapies. SIGNIFICANCE: This study suggests that ssDNA replication gaps are fundamental to the toxicity of genotoxic agents and underlie the BRCA-cancer phenotype "BRCAness," yielding promising biomarkers, targets, and opportunities to resensitize refractory disease.See related commentary by Canman, p. 1214.
©2020 American Association for Cancer Research.
Conflict of interest statement
CONFLICT OF INTERESTS
The authors declare no conflict of interests.
Figures
Comment in
-
Which Holds the Key to BRCAness: Inability to Repair the Break, Protect the Fork, or Prevent the Gap?Cancer Res. 2021 Mar 1;81(5):1214-1215. doi: 10.1158/0008-5472.CAN-20-4340. Cancer Res. 2021. PMID: 33822743
Comment on
-
Which Holds the Key to BRCAness: Inability to Repair the Break, Protect the Fork, or Prevent the Gap?Cancer Res. 2021 Mar 1;81(5):1214-1215. doi: 10.1158/0008-5472.CAN-20-4340. Cancer Res. 2021. PMID: 33822743
Similar articles
-
Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency.Mol Cell. 2021 Aug 5;81(15):3128-3144.e7. doi: 10.1016/j.molcel.2021.06.011. Epub 2021 Jul 2. Mol Cell. 2021. PMID: 34216544 Free PMC article.
-
Revisiting the BRCA-pathway through the lens of replication gap suppression: "Gaps determine therapy response in BRCA mutant cancer".DNA Repair (Amst). 2021 Nov;107:103209. doi: 10.1016/j.dnarep.2021.103209. Epub 2021 Aug 13. DNA Repair (Amst). 2021. PMID: 34419699 Free PMC article. Review.
-
BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks.Nature. 2014 Jun 26;510(7506):556-9. doi: 10.1038/nature13295. Epub 2014 Apr 28. Nature. 2014. PMID: 24776801 Free PMC article.
-
Therapeutic exploitation of tumor cell defects in homologous recombination.Anticancer Agents Med Chem. 2008 May;8(4):448-60. doi: 10.2174/187152008784220267. Anticancer Agents Med Chem. 2008. PMID: 18473729 Review.
-
(Single-stranded DNA) gaps in understanding BRCAness.Trends Genet. 2024 Sep;40(9):757-771. doi: 10.1016/j.tig.2024.04.013. Epub 2024 May 23. Trends Genet. 2024. PMID: 38789375 Review.
Cited by
-
Biochemical and structural basis for YTH domain of human YTHDC1 binding to methylated adenine in DNA.Nucleic Acids Res. 2020 Oct 9;48(18):10329-10341. doi: 10.1093/nar/gkaa604. Nucleic Acids Res. 2020. PMID: 32663306 Free PMC article.
-
Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency.Mol Cell. 2021 Aug 5;81(15):3128-3144.e7. doi: 10.1016/j.molcel.2021.06.011. Epub 2021 Jul 2. Mol Cell. 2021. PMID: 34216544 Free PMC article.
-
Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality.Cancers (Basel). 2022 Nov 24;14(23):5795. doi: 10.3390/cancers14235795. Cancers (Basel). 2022. PMID: 36497275 Free PMC article. Review.
-
Inhibitors of Rho kinases (ROCK) induce multiple mitotic defects and synthetic lethality in BRCA2-deficient cells.Elife. 2023 Apr 19;12:e80254. doi: 10.7554/eLife.80254. Elife. 2023. PMID: 37073955 Free PMC article.
-
CRISPR knockout genome-wide screens identify the HELQ-RAD52 axis in regulating the repair of cisplatin-induced single stranded DNA gaps.bioRxiv [Preprint]. 2024 Apr 20:2024.04.17.589988. doi: 10.1101/2024.04.17.589988. bioRxiv. 2024. Update in: Nucleic Acids Res. 2024 Dec 11;52(22):13832-13848. doi: 10.1093/nar/gkae998. PMID: 38659927 Free PMC article. Updated. Preprint.
References
-
- King MC et al., Genetic analysis of human breast cancer: literature review and description of family data in workshop. Genet Epidemiol Suppl 1, 3–13 (1986). - PubMed
-
- Munnell EW, The changing prognosis and treatment in cancer of the ovary. A report of 235 patients with primary ovarian carcinoma 1952–1961. Am J Obstet Gynecol 100, 790–805 (1968). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
