

Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations
- PMID: 33184173
- PMCID: PMC7720151
- DOI: 10.1073/pnas.2007840117
Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations
Abstract
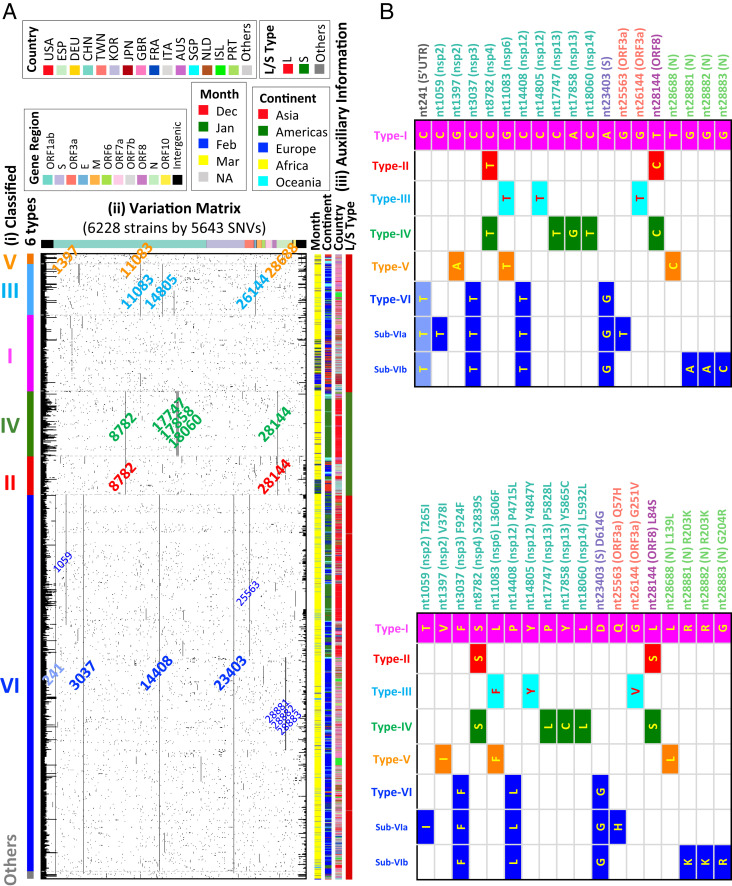
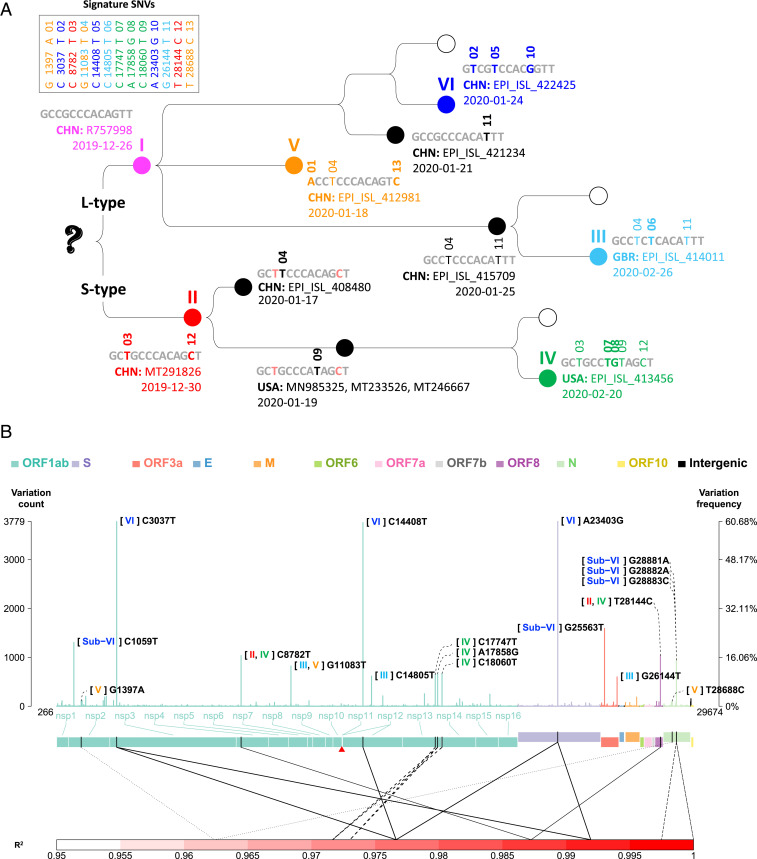
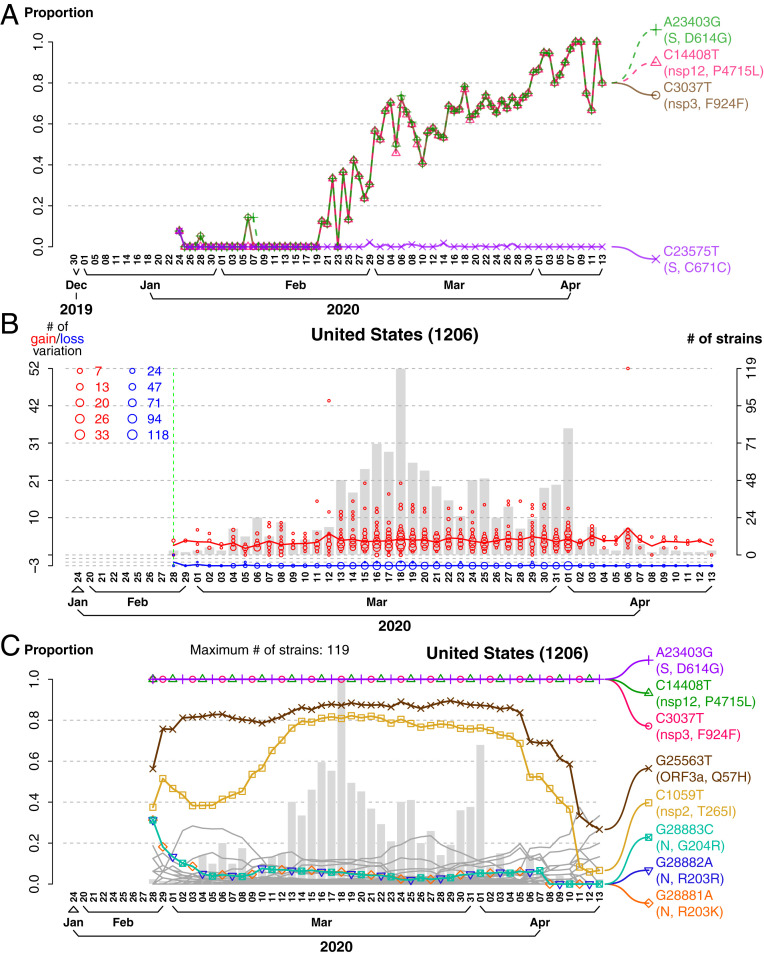
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causal agent of COVID 19, continues to evolve since its first emergence in December 2019. Using the complete sequences of 1,932 SARS-CoV-2 genomes, various clustering analyses consistently identified six types of the strains. Independent of the dendrogram construction, 13 signature variations in the form of single nucleotide variations (SNVs) in protein coding regions and one SNV in the 5' untranslated region (UTR) were identified and provided a direct interpretation for the six types (types I to VI). The six types of the strains and their underlying signature SNVs were validated in two subsequent analyses of 6,228 and 38,248 SARS-CoV-2 genomes which became available later. To date, type VI, characterized by the four signature SNVs C241T (5'UTR), C3037T (nsp3 F924F), C14408T (nsp12 P4715L), and A23403G (Spike D614G), with strong allelic associations, has become the dominant type. Since C241T is in the 5' UTR with uncertain significance and the characteristics can be captured by the other three strongly associated SNVs, we focus on the other three. The increasing frequency of the type VI haplotype 3037T-14408T-23403G in the majority of the submitted samples in various countries suggests a possible fitness gain conferred by the type VI signature SNVs. The fact that strains missing one or two of these signature SNVs fail to persist implies possible interactions among these SNVs. Later SNVs such as G28881A, G28882A, and G28883C have emerged with strong allelic associations, forming new subtypes. This study suggests that SNVs may become an important consideration in SARS-CoV-2 classification and surveillance.
Keywords: COVID-19; allelic association; mutation; sequencing; single nucleotide variation.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures

References
-
- Sokal R. R., Michener C. D., A statistical method for evaluating systematic relationships. Univ. Kansas Sci. Bull. 28, 1409−1438 (1958).
-
- Saitou N., Nei M., The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987). - PubMed
-
- Felsenstein J., Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981). - PubMed
-
- Edwards A. W. F., Cavallisforza L. L., The reconstruction of evolution. Ann. Hum. Genet. 27, 104–105 (1963).
-
- Everitt B. S., Landau S., Leese M., Cluster Analysis (Arnold, London, United Kingdom, 2001).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
