

Markov state modeling reveals alternative unbinding pathways for peptide-MHC complexes
- PMID: 33184174
- PMCID: PMC7720115
- DOI: 10.1073/pnas.2007246117
Markov state modeling reveals alternative unbinding pathways for peptide-MHC complexes
Abstract
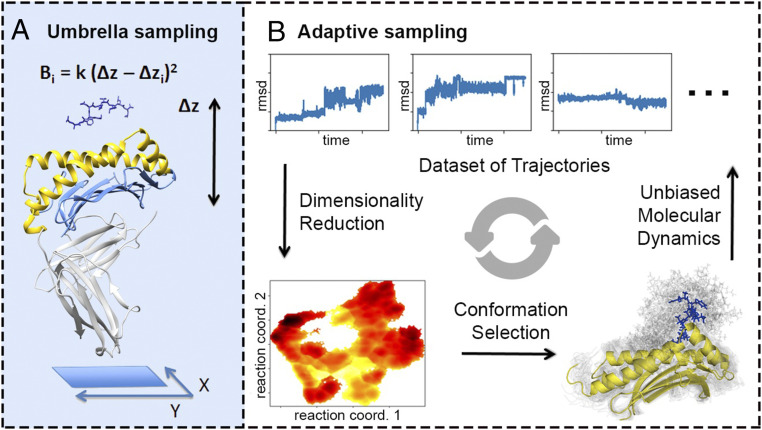
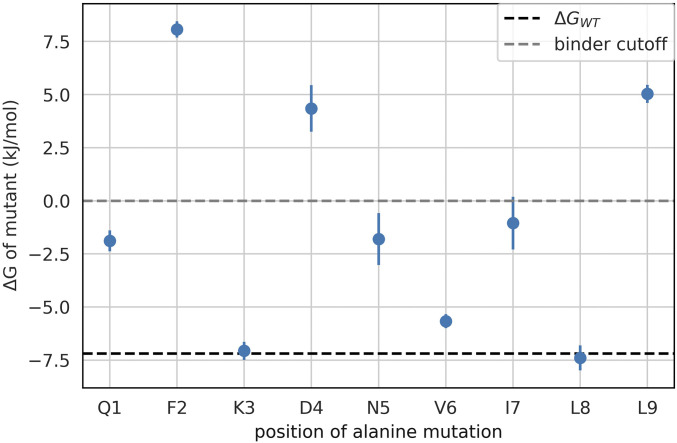
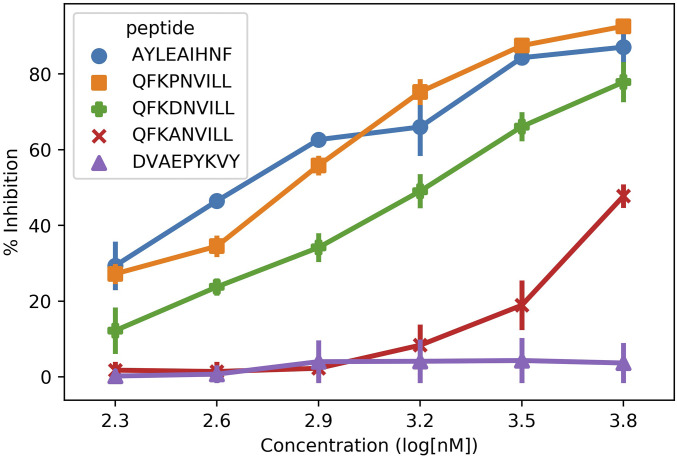
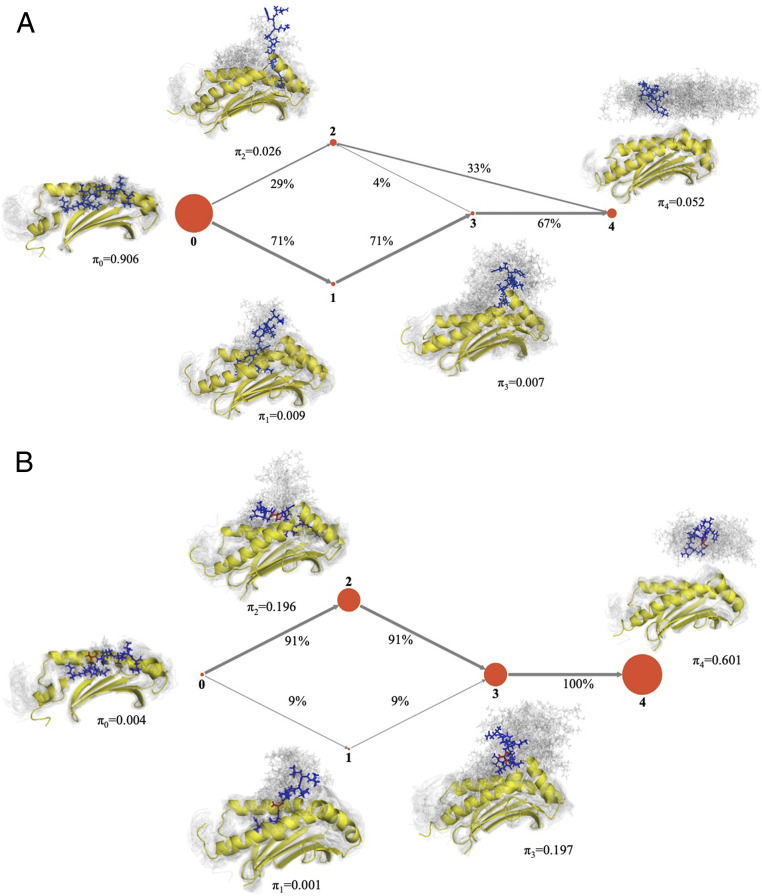
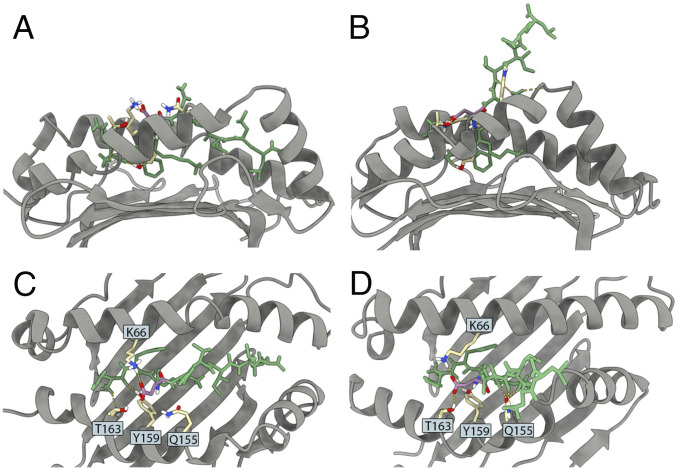
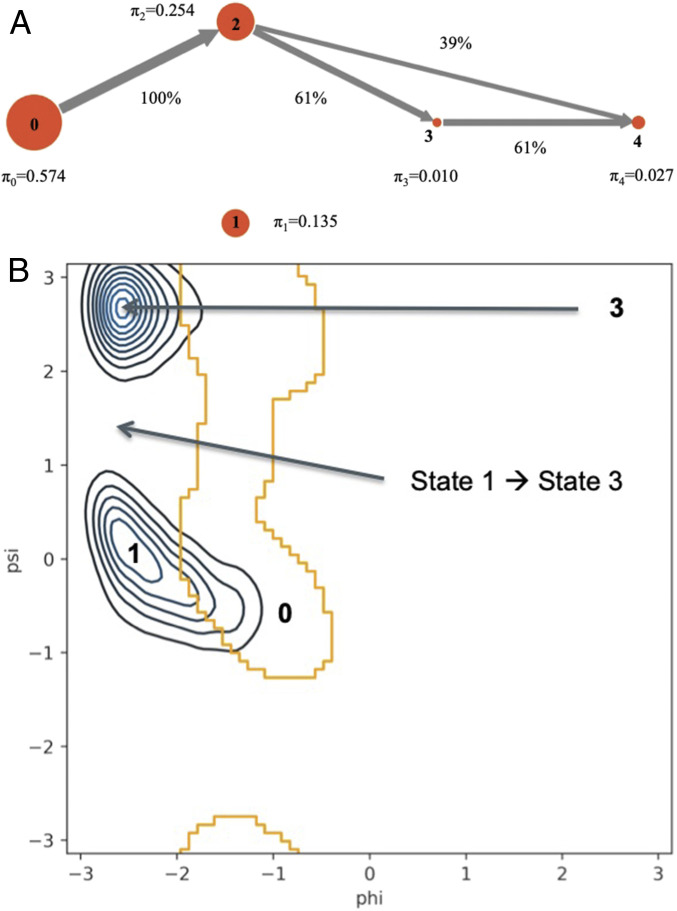
Peptide binding to major histocompatibility complexes (MHCs) is a central component of the immune system, and understanding the mechanism behind stable peptide-MHC binding will aid the development of immunotherapies. While MHC binding is mostly influenced by the identity of the so-called anchor positions of the peptide, secondary interactions from nonanchor positions are known to play a role in complex stability. However, current MHC-binding prediction methods lack an analysis of the major conformational states and might underestimate the impact of secondary interactions. In this work, we present an atomically detailed analysis of peptide-MHC binding that can reveal the contributions of any interaction toward stability. We propose a simulation framework that uses both umbrella sampling and adaptive sampling to generate a Markov state model (MSM) for a coronavirus-derived peptide (QFKDNVILL), bound to one of the most prevalent MHC receptors in humans (HLA-A24:02). While our model reaffirms the importance of the anchor positions of the peptide in establishing stable interactions, our model also reveals the underestimated importance of position 4 (p4), a nonanchor position. We confirmed our results by simulating the impact of specific peptide mutations and validated these predictions through competitive binding assays. By comparing the MSM of the wild-type system with those of the D4A and D4P mutations, our modeling reveals stark differences in unbinding pathways. The analysis presented here can be applied to any peptide-MHC complex of interest with a structural model as input, representing an important step toward comprehensive modeling of the MHC class I pathway.
Keywords: Markov state modeling; adaptive sampling; competitive binding assay; peptide–MHC binding stability.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Changes at the floor of the peptide-binding groove induce a strong preference for proline at position 3 of the bound peptide: molecular dynamics simulations of HLA-A*0217.Biopolymers. 2000 Oct 15;54(5):318-27. doi: 10.1002/1097-0282(20001015)54:5<318::AID-BIP30>3.0.CO;2-T. Biopolymers. 2000. PMID: 10935972
-
Predicting sequences and structures of MHC-binding peptides: a computational combinatorial approach.J Comput Aided Mol Des. 2001 Jun;15(6):573-86. doi: 10.1023/a:1011145123635. J Comput Aided Mol Des. 2001. PMID: 11495228
-
MHC class I/peptide interactions: binding specificity and kinetics.J Mol Recognit. 1993 Jun;6(2):59-69. doi: 10.1002/jmr.300060204. J Mol Recognit. 1993. PMID: 8305252 Review.
-
Application of genetic search in derivation of matrix models of peptide binding to MHC molecules.Proc Int Conf Intell Syst Mol Biol. 1997;5:75-83. Proc Int Conf Intell Syst Mol Biol. 1997. PMID: 9322018
-
Non-canonical peptides bound to MHC.Curr Pharm Des. 2009;15(28):3274-82. doi: 10.2174/138161209789105090. Curr Pharm Des. 2009. PMID: 19860676 Review.
Cited by
-
Mutations in glioblastoma proteins do not disrupt epitope presentation and recognition, maintaining a specific CD8 T cell immune response potential.Sci Rep. 2024 Jul 19;14(1):16721. doi: 10.1038/s41598-024-67099-2. Sci Rep. 2024. PMID: 39030304 Free PMC article.
-
From Data to Knowledge: Systematic Review of Tools for Automatic Analysis of Molecular Dynamics Output.Front Pharmacol. 2022 Mar 10;13:844293. doi: 10.3389/fphar.2022.844293. eCollection 2022. Front Pharmacol. 2022. PMID: 35359865 Free PMC article. Review.
-
Loading dynamics of one SARS-CoV-2-derived peptide into MHC-II revealed by kinetic models.Biophys J. 2023 May 2;122(9):1665-1677. doi: 10.1016/j.bpj.2023.03.032. Epub 2023 Mar 24. Biophys J. 2023. PMID: 36964657 Free PMC article.
-
EnGens: a computational framework for generation and analysis of representative protein conformational ensembles.bioRxiv [Preprint]. 2023 Apr 28:2023.04.24.538094. doi: 10.1101/2023.04.24.538094. bioRxiv. 2023. Update in: Brief Bioinform. 2023 Jul 20;24(4):bbad242. doi: 10.1093/bib/bbad242. PMID: 37163076 Free PMC article. Updated. Preprint.
-
APE-Gen2.0: Expanding Rapid Class I Peptide-Major Histocompatibility Complex Modeling to Post-Translational Modifications and Noncanonical Peptide Geometries.J Chem Inf Model. 2024 Mar 11;64(5):1730-1750. doi: 10.1021/acs.jcim.3c01667. Epub 2024 Feb 28. J Chem Inf Model. 2024. PMID: 38415656 Free PMC article.
References
-
- Harndahl M., et al. , Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur. J. Immunol. 42, 1405–1416 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
