

A human cell atlas of fetal chromatin accessibility
- PMID: 33184180
- PMCID: PMC7785298
- DOI: 10.1126/science.aba7612
A human cell atlas of fetal chromatin accessibility
Abstract
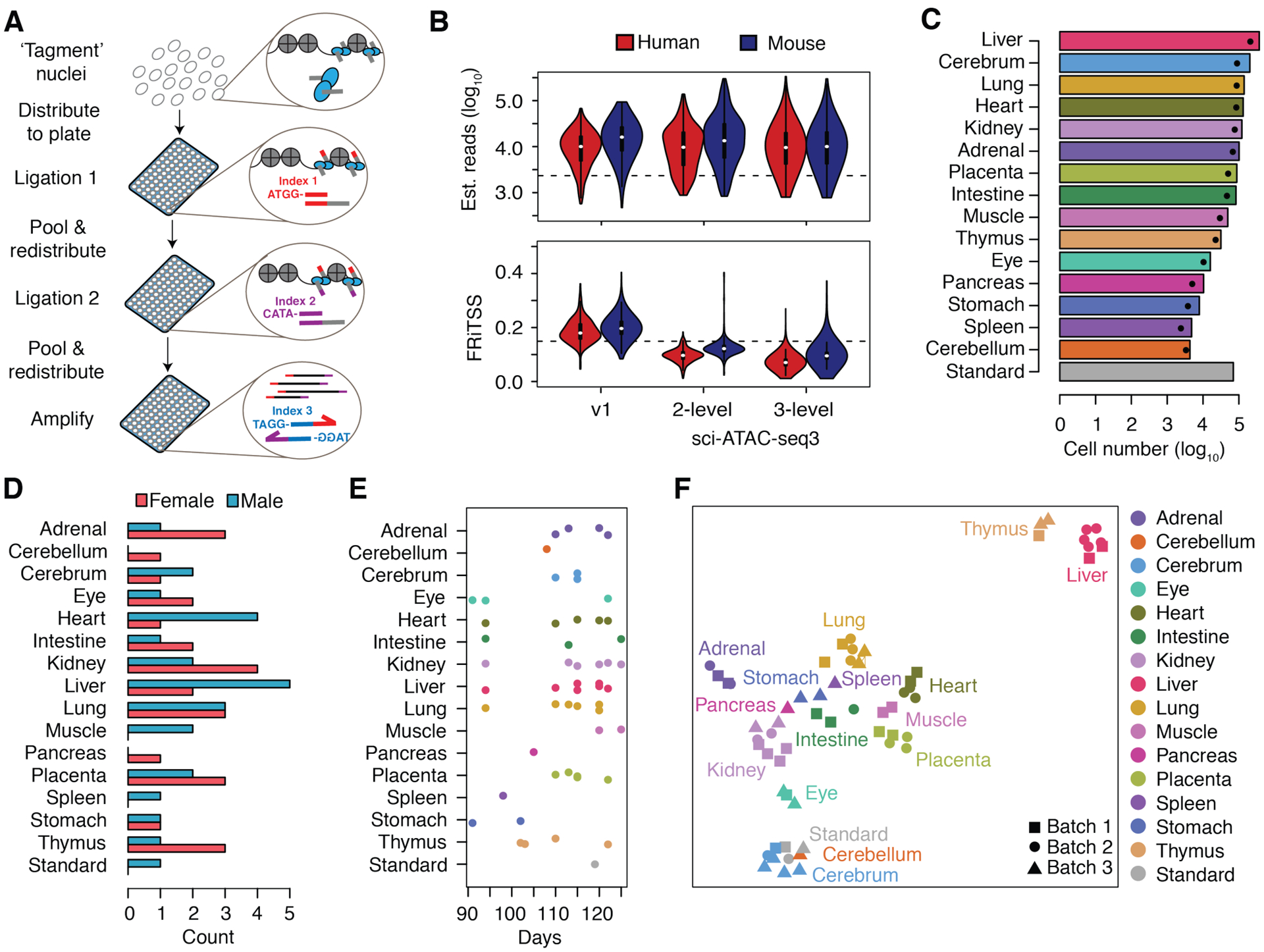
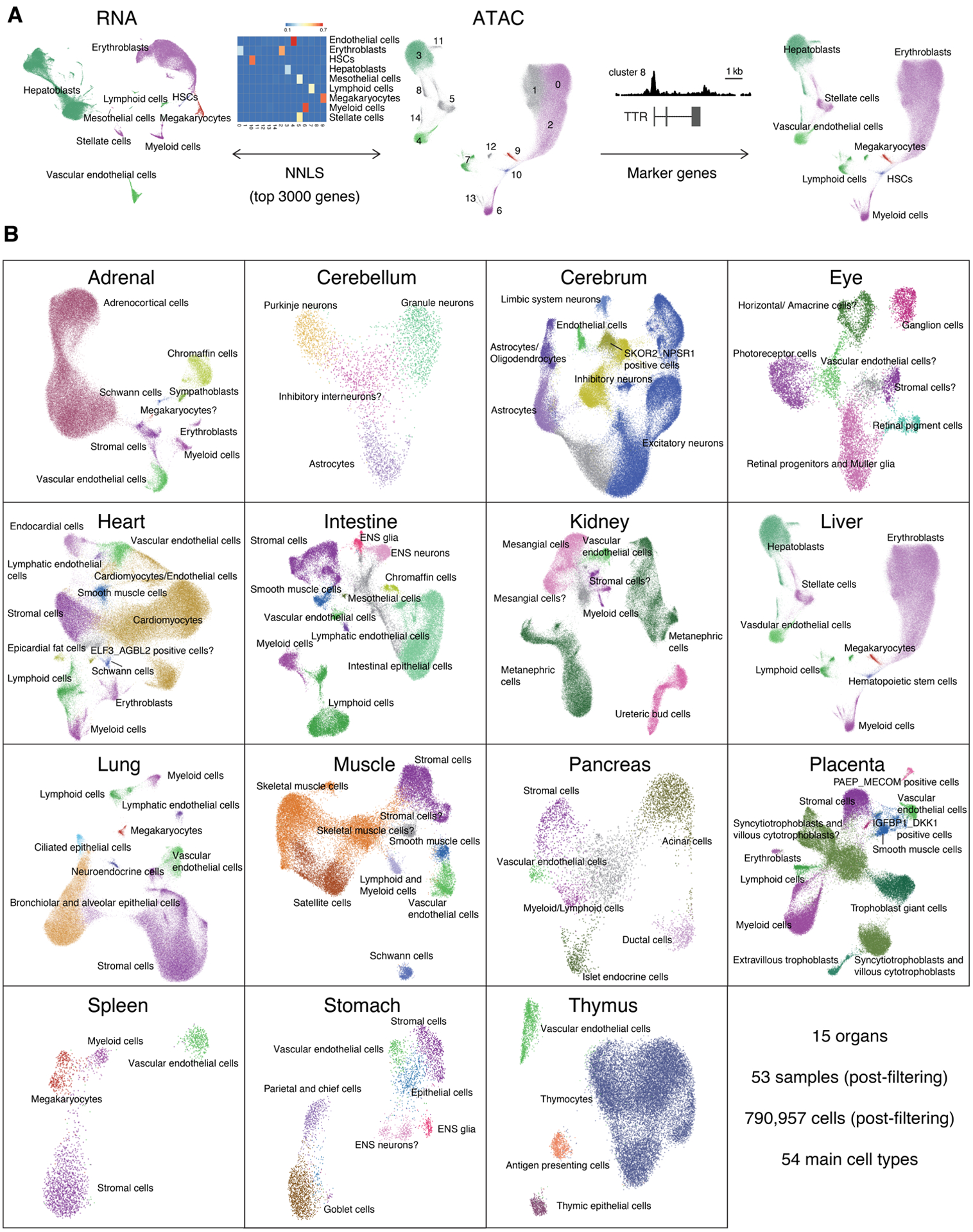
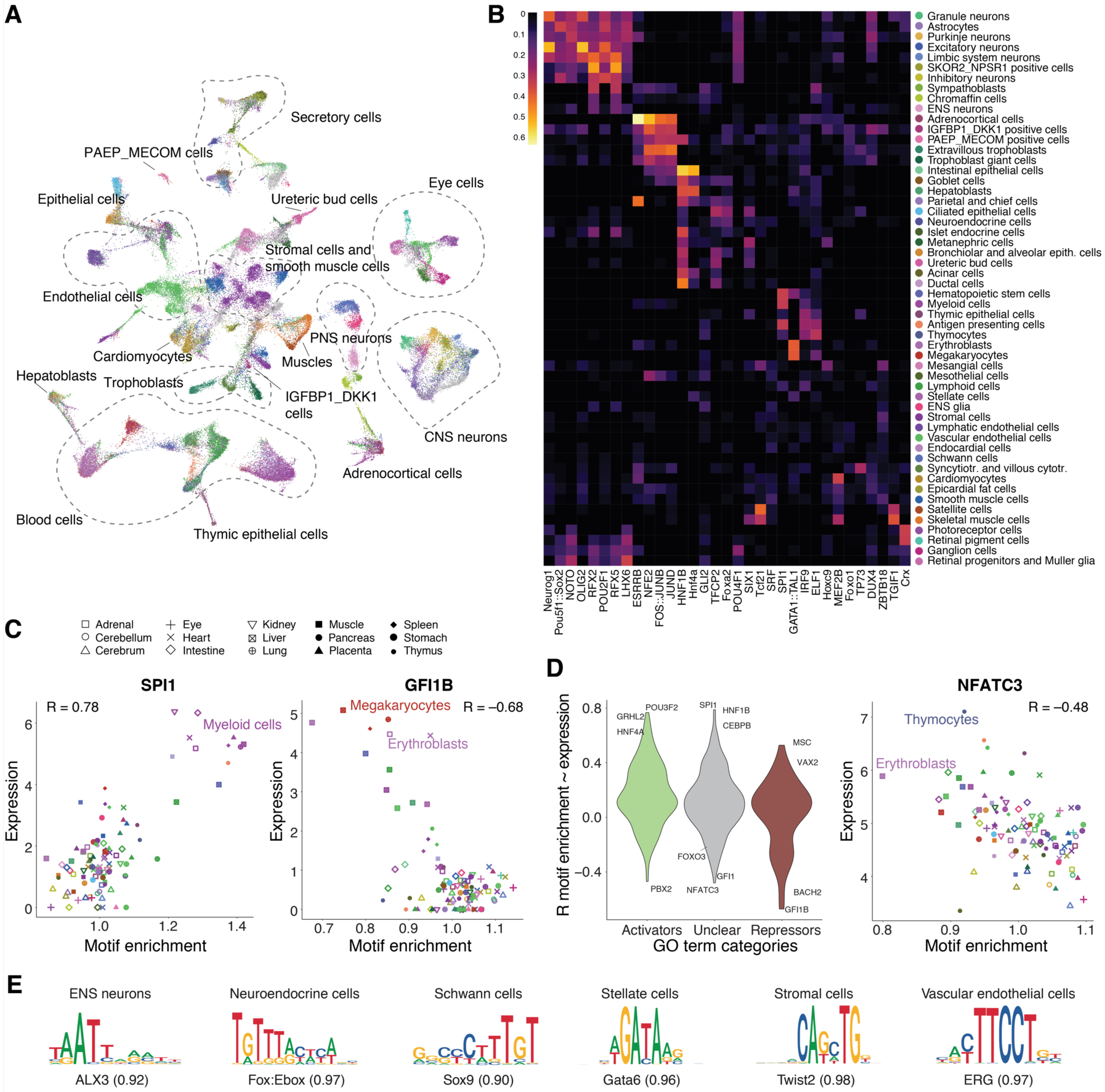
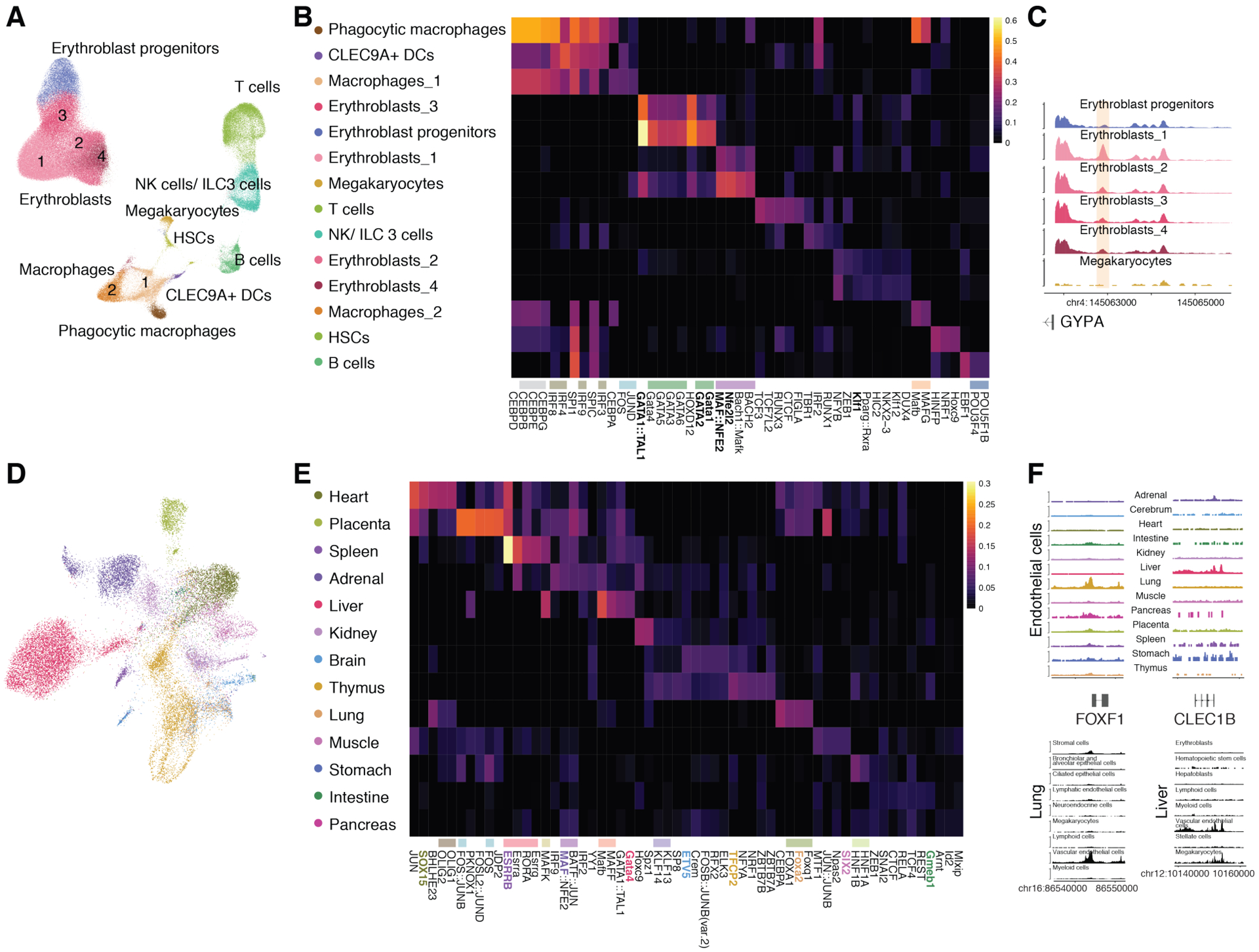
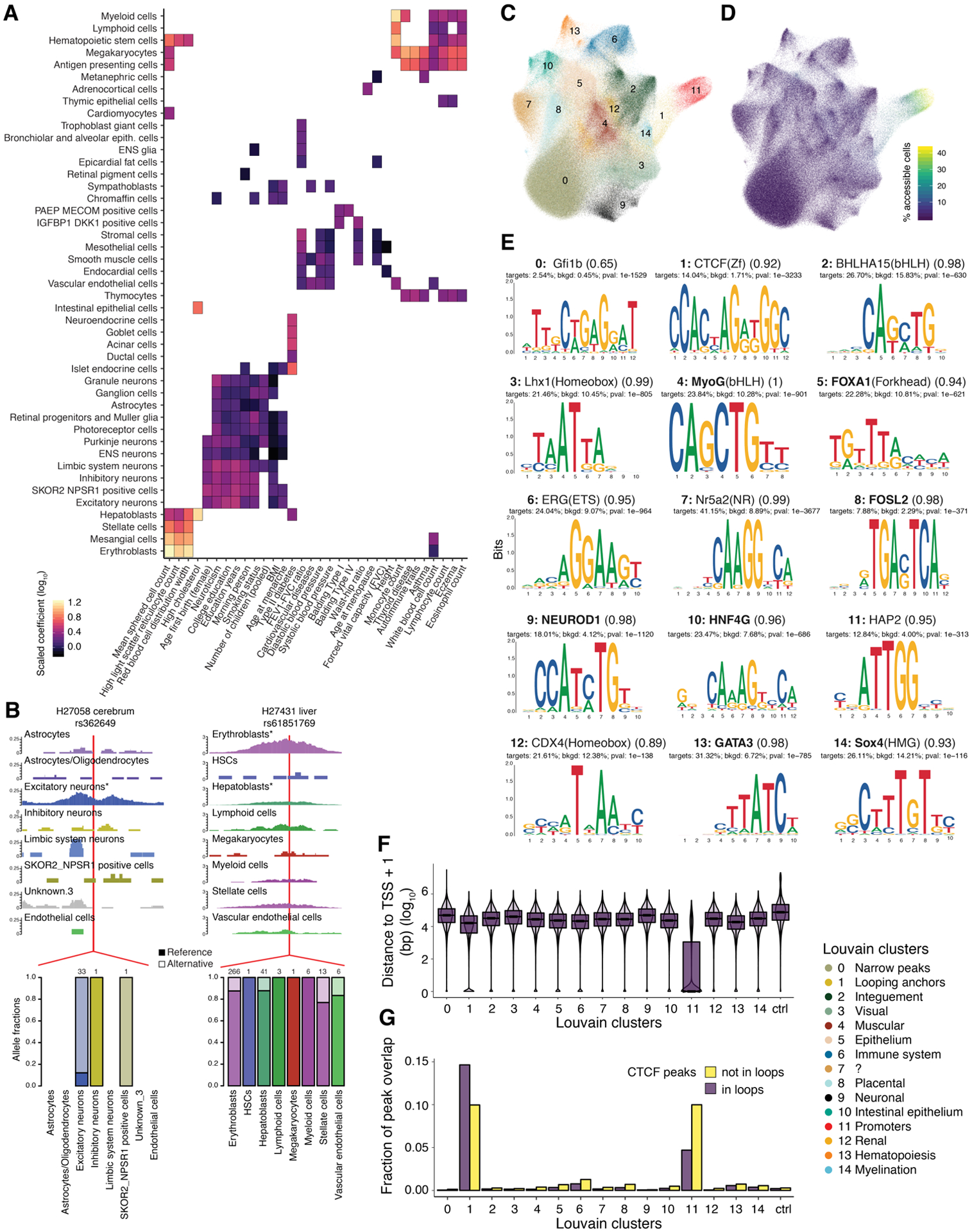
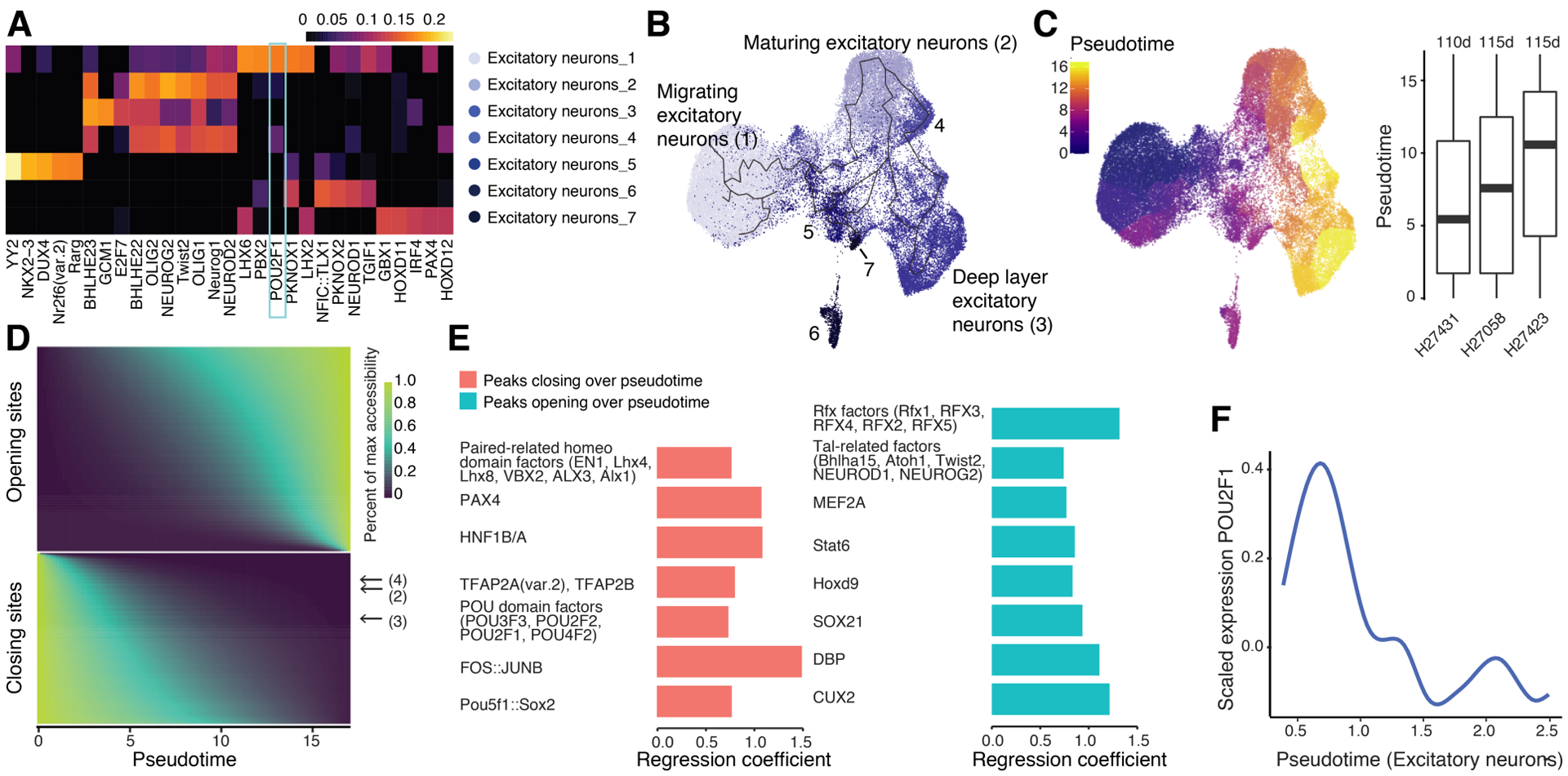
The chromatin landscape underlying the specification of human cell types is of fundamental interest. We generated human cell atlases of chromatin accessibility and gene expression in fetal tissues. For chromatin accessibility, we devised a three-level combinatorial indexing assay and applied it to 53 samples representing 15 organs, profiling ~800,000 single cells. We leveraged cell types defined by gene expression to annotate these data and cataloged hundreds of thousands of candidate regulatory elements that exhibit cell type-specific chromatin accessibility. We investigated the properties of lineage-specific transcription factors (such as POU2F1 in neurons), organ-specific specializations of broadly distributed cell types (such as blood and endothelial), and cell type-specific enrichments of complex trait heritability. These data represent a rich resource for the exploration of in vivo human gene regulation in diverse tissues and cell types.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
A molecular handbook for human development.Nature. 2021 Feb;590(7844):43-44. doi: 10.1038/d41586-021-00142-8. Nature. 2021. PMID: 33500582 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
