

Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis
- PMID: 33184454
- PMCID: PMC7665074
- DOI: 10.1038/s41598-020-76881-x
Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis
Abstract
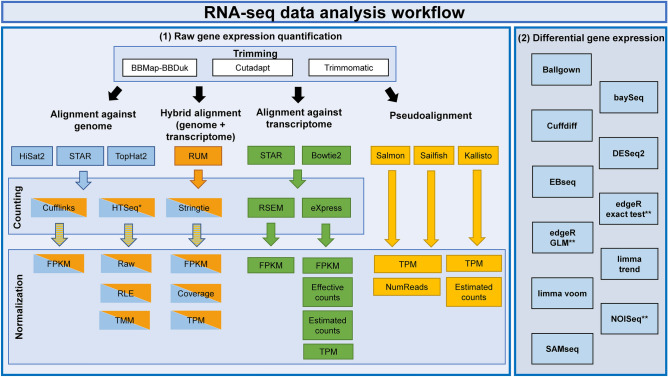
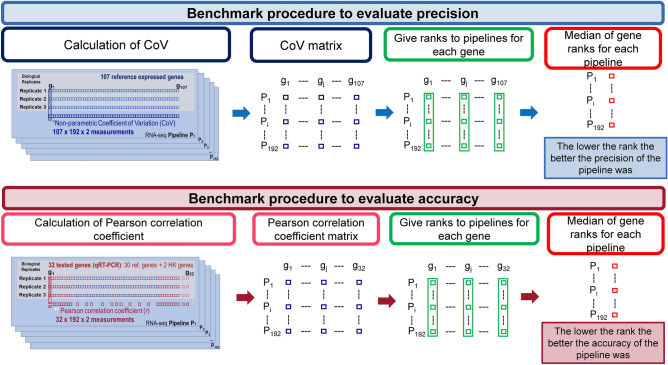
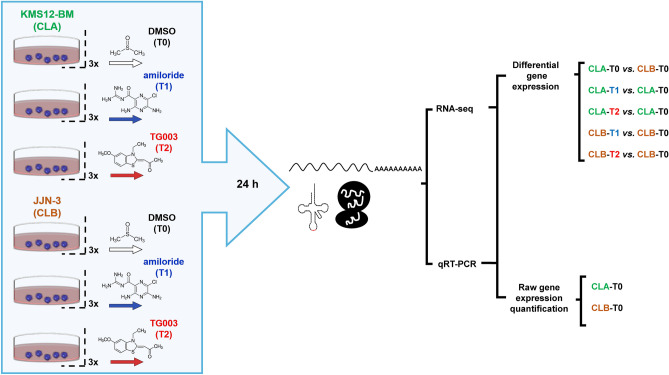
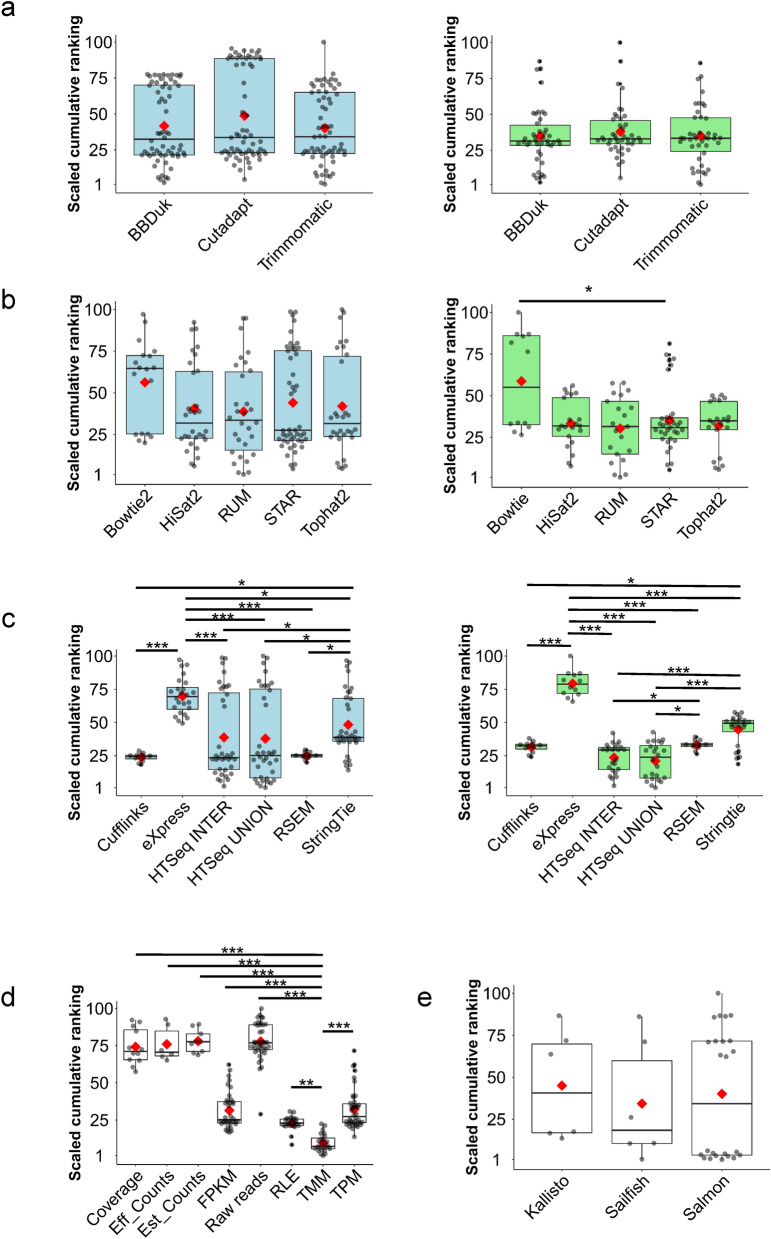
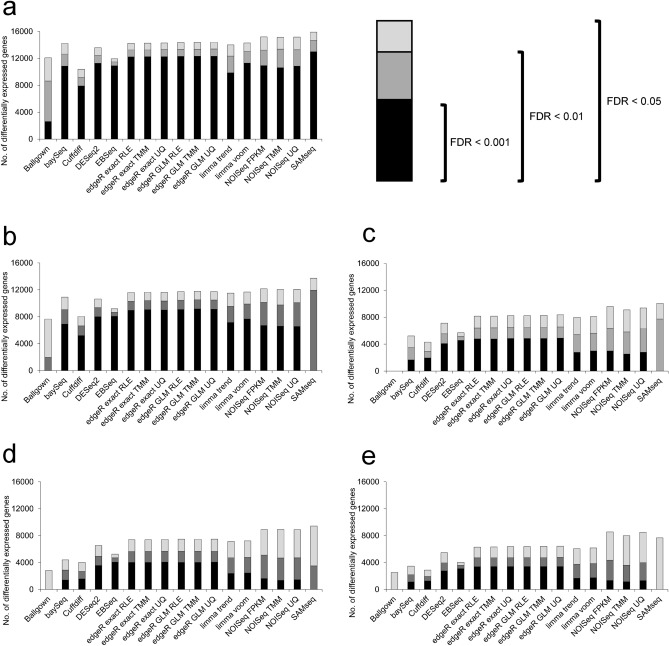
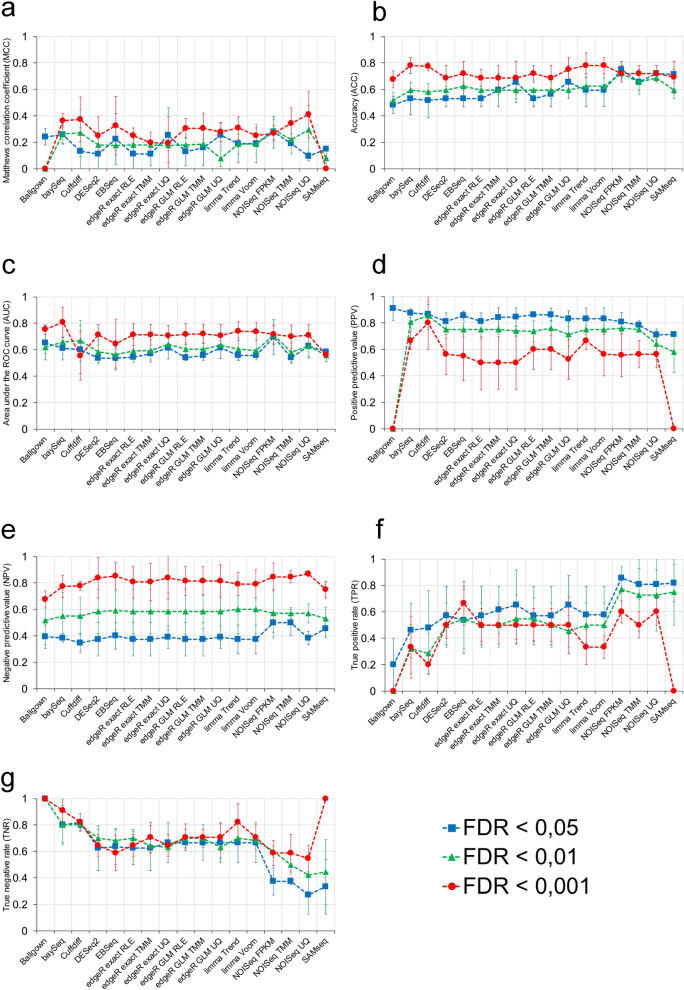
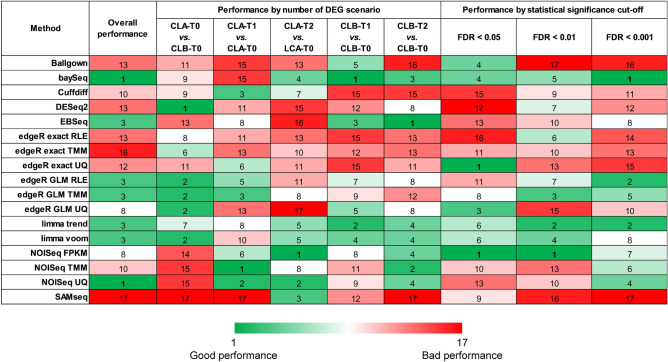
RNA-seq is currently considered the most powerful, robust and adaptable technique for measuring gene expression and transcription activation at genome-wide level. As the analysis of RNA-seq data is complex, it has prompted a large amount of research on algorithms and methods. This has resulted in a substantial increase in the number of options available at each step of the analysis. Consequently, there is no clear consensus about the most appropriate algorithms and pipelines that should be used to analyse RNA-seq data. In the present study, 192 pipelines using alternative methods were applied to 18 samples from two human cell lines and the performance of the results was evaluated. Raw gene expression signal was quantified by non-parametric statistics to measure precision and accuracy. Differential gene expression performance was estimated by testing 17 differential expression methods. The procedures were validated by qRT-PCR in the same samples. This study weighs up the advantages and disadvantages of the tested algorithms and pipelines providing a comprehensive guide to the different methods and procedures applied to the analysis of RNA-seq data, both for the quantification of the raw expression signal and for the differential gene expression.
Conflict of interest statement
N.C.G. Honoraria: Janssen. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
