

Re-engineering Antimicrobial Peptides into Oncolytics Targeting Drug-Resistant Ovarian Cancers
- PMID: 33184577
- PMCID: PMC7596148
- DOI: 10.1007/s12195-020-00626-z
Re-engineering Antimicrobial Peptides into Oncolytics Targeting Drug-Resistant Ovarian Cancers
Abstract
Introduction: Bacteria and cancer cells share a common trait-both possess an electronegative surface that distinguishes them from healthy mammalian counterparts. This opens opportunities to repurpose antimicrobial peptides (AMPs), which are cationic amphiphiles that kill bacteria by disrupting their anionic cell envelope, into anticancer peptides (ACPs). To test this assertion, we investigate the mechanisms by which a pathogen-specific AMP, originally designed to kill bacterial Tuberculosis, potentiates the lytic destruction of drug-resistant cancers and synergistically enhances chemotherapeutic potency.
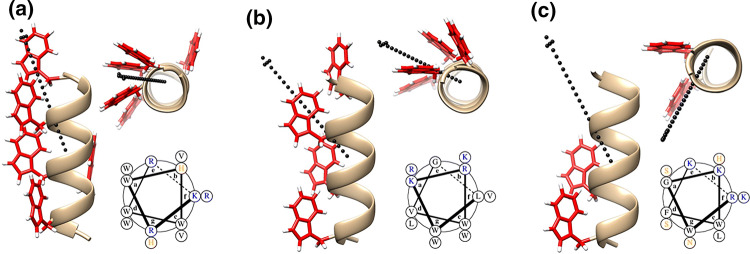
Materials and methods: De novo peptide design, paired with cellular assays, elucidate structure-activity relationships (SAR) important to ACP potency and specificity. Using the sequence MAD1, microscopy, spectrophotometry and flow cytometry identify the peptide's anticancer mechanisms, while parallel combinatorial screens define chemotherapeutic synergy in drug-resistant cell lines and patient derived ex vivo tumors.
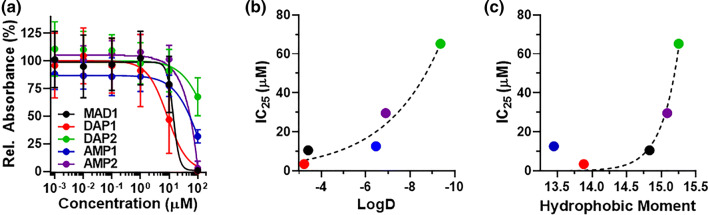
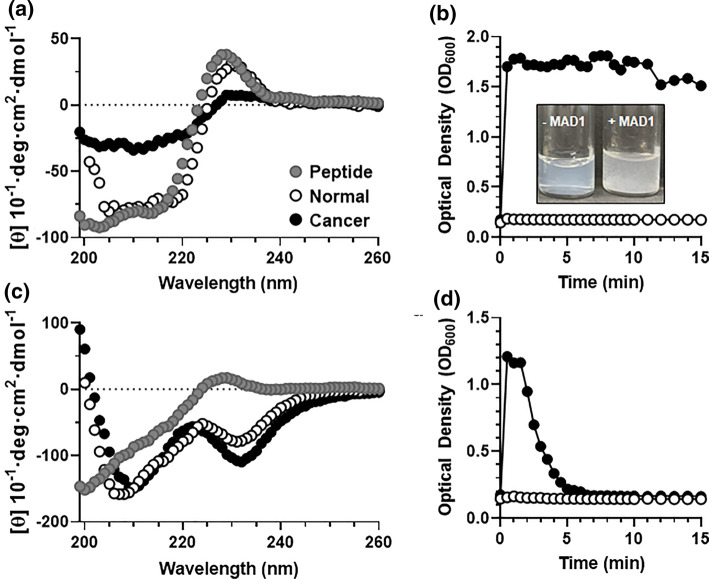
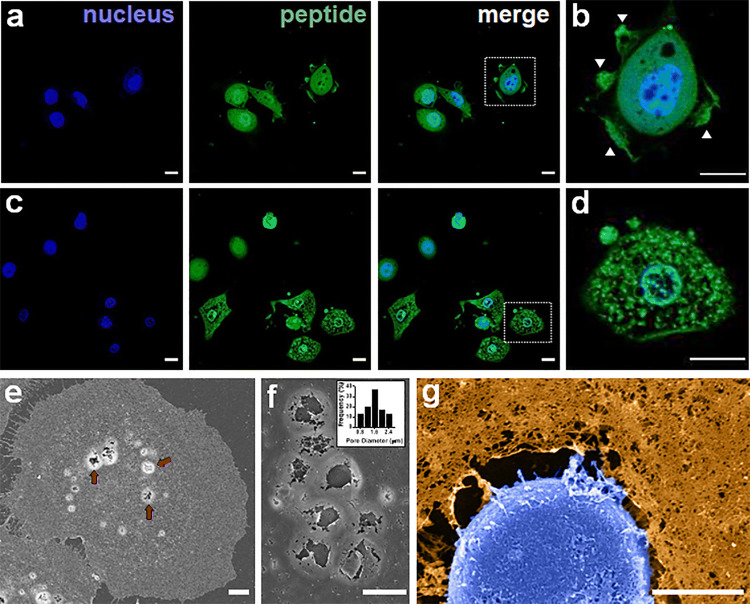
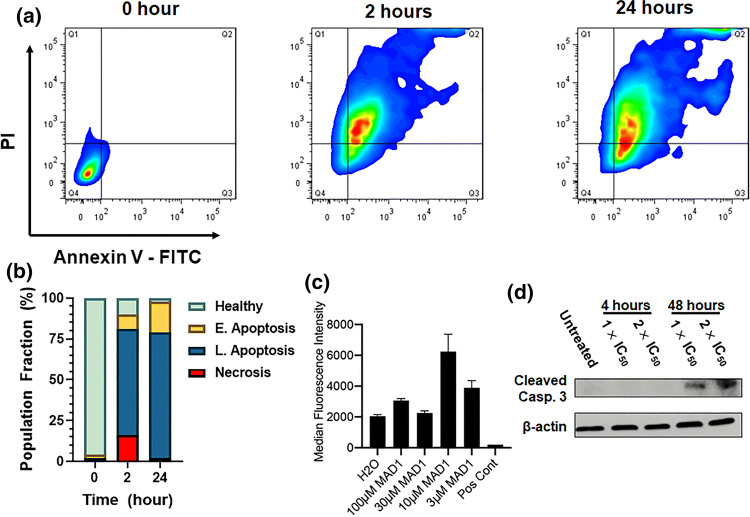
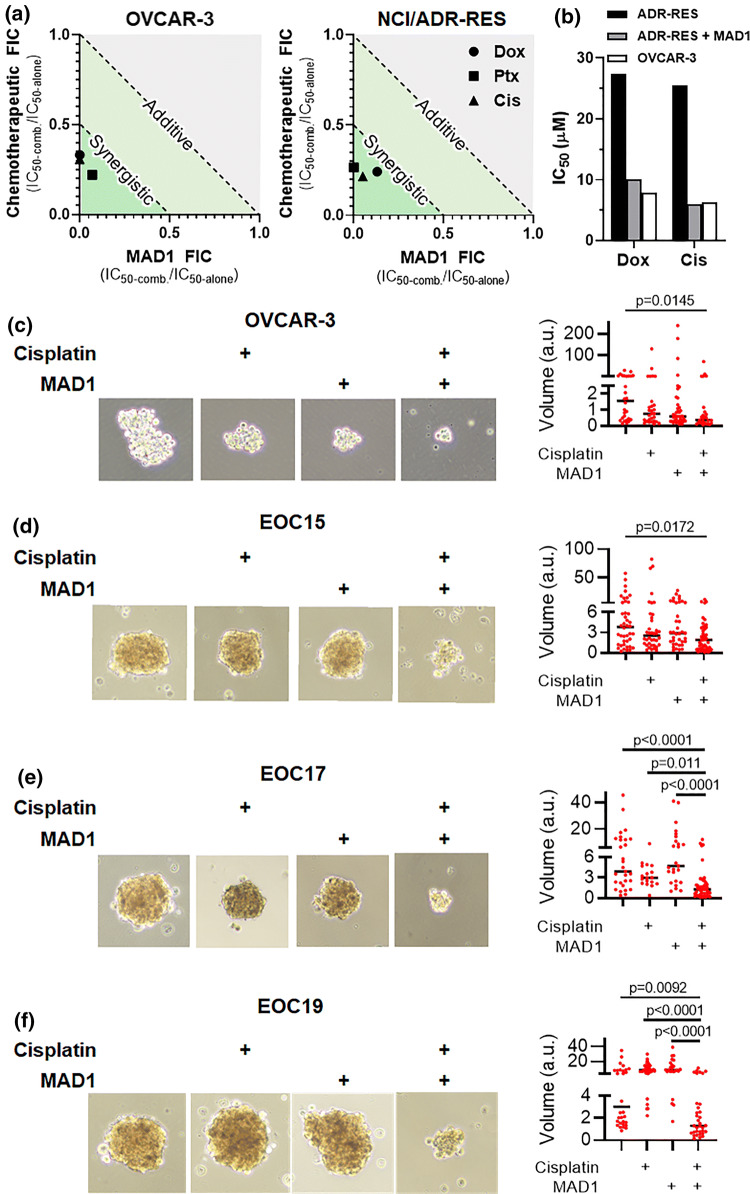
Results: SAR investigations reveal spatial sequestration of amphiphilic regions increases ACP potency, but at the cost of specificity. Selecting MAD1 as a lead sequence, mechanistic studies identify that the peptide forms pore-like supramolecular assemblies within the plasma and nuclear membranes of cancer cells to potentiate death through lytic and apoptotic mechanisms. This diverse activity enables MAD1 to synergize broadly with chemotherapeutics, displaying remarkable combinatorial efficacy against drug-resistant ovarian carcinoma cells and patient-derived tumor spheroids.
Conclusions: We show that cancer-specific ACPs can be rationally engineered using nature's AMP toolbox as templates. Selecting the antimicrobial peptide MAD1, we demonstrate the potential of this strategy to open a wealth of synthetic biotherapies that offer new, combinatorial opportunities against drug resistant tumors.
Keywords: Anticancer peptides; Combinatorial therapy; De novo design; Nanostructures; Supramolecular assembly.
© Biomedical Engineering Society 2020.
Figures
References
-
- Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer. 2003;3:502–516. - PubMed
-
- Aina OH, Liu R, Sutcliffe JL, Marik J, Pan C-X, Lam KS. From combinatorial chemistry to cancer-targeting peptides. Mol. Pharm. 2007;4:631–651. - PubMed
-
- Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012;30:679. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
