

On the faithfulness of molecular mechanics representations of proteins towards quantum-mechanical energy surfaces
- PMID: 33184586
- PMCID: PMC7653345
- DOI: 10.1098/rsfs.2019.0121
On the faithfulness of molecular mechanics representations of proteins towards quantum-mechanical energy surfaces
Abstract
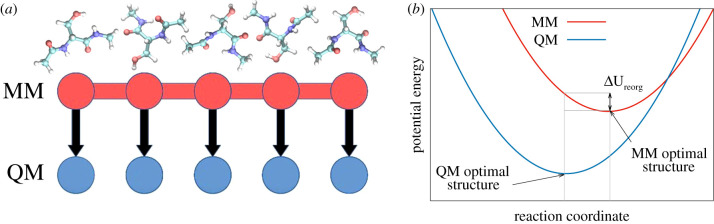
Force fields based on molecular mechanics (MM) are the main computational tool to study the relationship between protein structure and function at the molecular level. To validate the quality of such force fields, high-level quantum-mechanical (QM) data are employed to test their capability to reproduce the features of all major conformational substates of a series of blocked amino acids. The phase-space overlap between MM and QM is quantified in terms of the average structural reorganization energies over all energy minima. Here, the structural reorganization energy is the MM potential-energy difference between the structure of the respective QM energy minimum and the structure of the closest MM energy minimum. Thus, it serves as a measure for the relative probability of visiting the QM minimum during an MM simulation. We evaluate variants of the AMBER, CHARMM, GROMOS and OPLS biomolecular force fields. In addition, the two blocked amino acids alanine and serine are used to demonstrate the dependence of the measured agreement on the QM method, the phase, and the conformational preferences. Blocked serine serves as an example to discuss possible improvements of the force fields, such as including polarization with Drude particles, or using tailored force fields. The results show that none of the evaluated force fields satisfactorily reproduces all energy minima. By decomposing the average structural reorganization energies in terms of individual energy terms, we can further assess the individual weaknesses of the parametrization strategies of each force field. The dominant problem for most force fields appears to be the van der Waals parameters, followed to a lesser degree by dihedral and bonded terms. Our results show that performing a simple QM energy optimization from an MM-optimized structure can be a first test of the validity of a force field for a particular target molecule.
Keywords: AMBER; CHARMM; GROMOS; OPLS; molecular mechanics; quantum mechanics.
© 2020 The Author(s).
Conflict of interest statement
We declare we have no competing interest.
Figures
Similar articles
-
Toward a general neural network force field for protein simulations: Refining the intramolecular interaction in protein.J Chem Phys. 2023 Jul 14;159(2):024118. doi: 10.1063/5.0142280. J Chem Phys. 2023. PMID: 37431910 Free PMC article.
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
-
Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations.J Comput Chem. 2004 Aug;25(11):1400-15. doi: 10.1002/jcc.20065. J Comput Chem. 2004. PMID: 15185334
-
Parametrization of Force Field Bonded Terms under Structural Inconsistency.J Chem Inf Model. 2022 Oct 10;62(19):4771-4782. doi: 10.1021/acs.jcim.2c00950. Epub 2022 Sep 16. J Chem Inf Model. 2022. PMID: 36112364
-
Force field development phase II: Relaxation of physics-based criteria… or inclusion of more rigorous physics into the representation of molecular energetics.J Comput Aided Mol Des. 2019 Feb;33(2):205-264. doi: 10.1007/s10822-018-0134-x. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506159 Review.
Cited by
-
Does the inclusion of electronic polarisability lead to a better modelling of peptide aggregation?RSC Adv. 2022 Jul 21;12(32):20829-20837. doi: 10.1039/d2ra01478e. eCollection 2022 Jul 14. RSC Adv. 2022. PMID: 35919139 Free PMC article.
-
Toward a general neural network force field for protein simulations: Refining the intramolecular interaction in protein.J Chem Phys. 2023 Jul 14;159(2):024118. doi: 10.1063/5.0142280. J Chem Phys. 2023. PMID: 37431910 Free PMC article.
-
On-Surface Synthesis and Cryogenic Exfoliation of Sterically Frustrated Atropisomers.ACS Nano. 2025 Apr 15;19(14):13805-13816. doi: 10.1021/acsnano.4c16645. Epub 2025 Apr 1. ACS Nano. 2025. PMID: 40168186 Free PMC article.
-
Integration of Experimental Data and Use of Automated Fitting Methods in Developing Protein Force Fields.Commun Chem. 2022;5:38. doi: 10.1038/s42004-022-00653-z. Epub 2022 Mar 18. Commun Chem. 2022. PMID: 35382231 Free PMC article. Review.
-
Transferable Classical Force Field for Pure and Mixed Metal Halide Perovskites Parameterized from First-Principles.J Chem Inf Model. 2022 Dec 26;62(24):6423-6435. doi: 10.1021/acs.jcim.1c01506. Epub 2022 May 16. J Chem Inf Model. 2022. PMID: 35576452 Free PMC article.
References
LinkOut - more resources
Full Text Sources
