

A clade of SARS-CoV-2 viruses associated with lower viral loads in patient upper airways
- PMID: 33186810
- PMCID: PMC7655495
- DOI: 10.1016/j.ebiom.2020.103112
A clade of SARS-CoV-2 viruses associated with lower viral loads in patient upper airways
Abstract
Background: The rapid spread of SARS-CoV-2, the causative agent of Coronavirus disease 2019 (COVID-19), has been accompanied by the emergence of distinct viral clades, though their clinical significance remains unclear. Here, we aimed to investigate the phylogenetic characteristics of SARS-CoV-2 infections in Chicago, Illinois, and assess their relationship to clinical parameters.
Methods: We performed whole-genome sequencing of SARS-CoV-2 isolates collected from COVID-19 patients in Chicago in mid-March, 2020. Using these and other publicly available sequences, we performed phylogenetic, phylogeographic, and phylodynamic analyses. Patient data was assessed for correlations between demographic or clinical characteristics and virologic features.
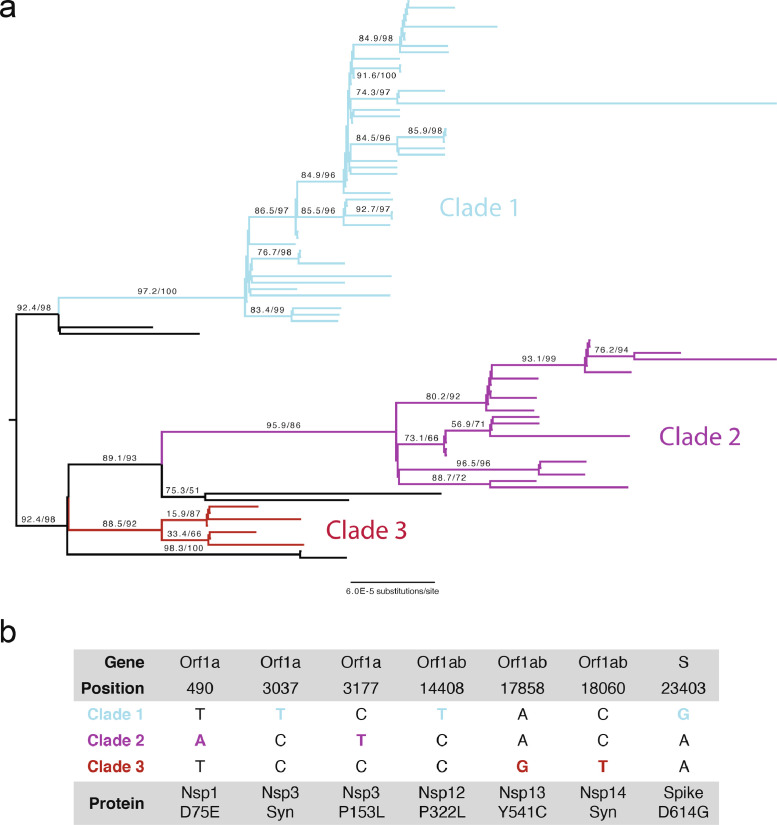
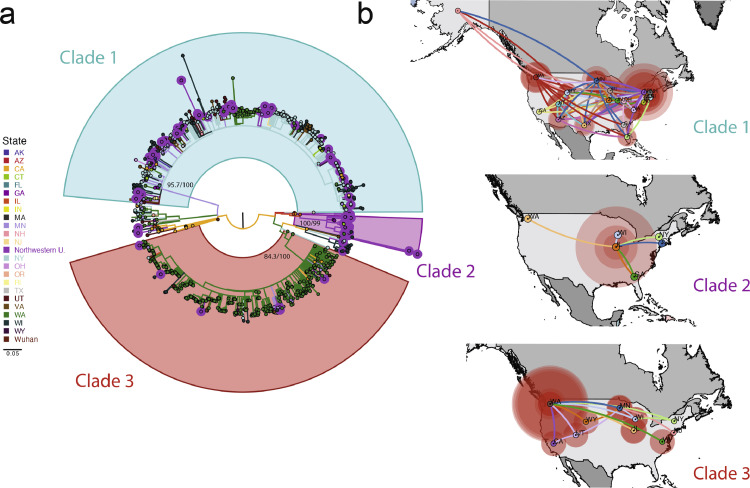
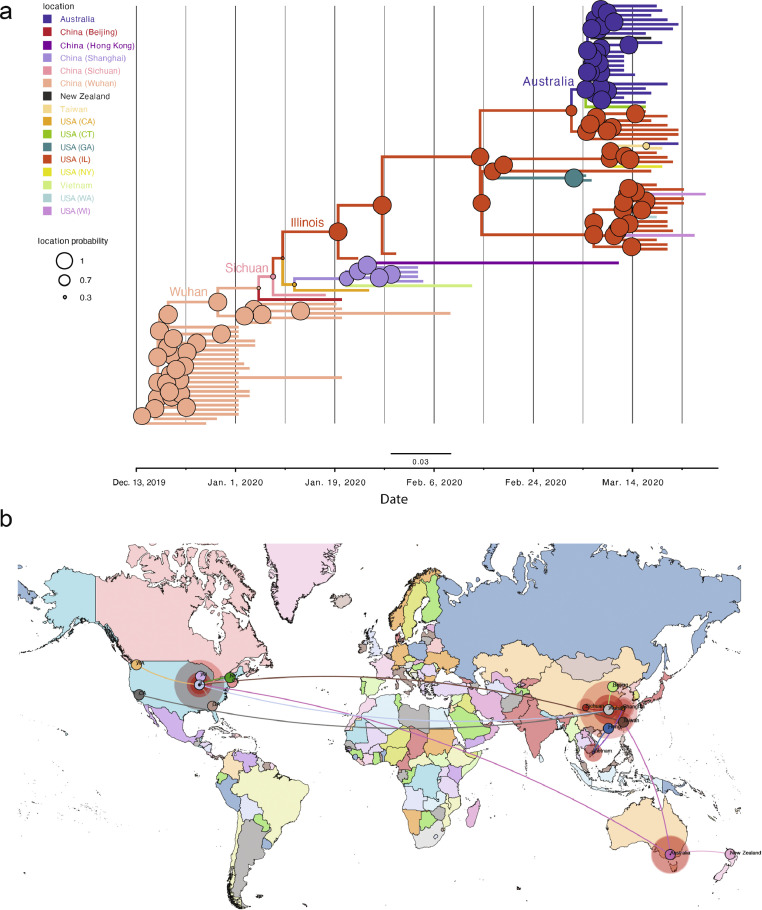
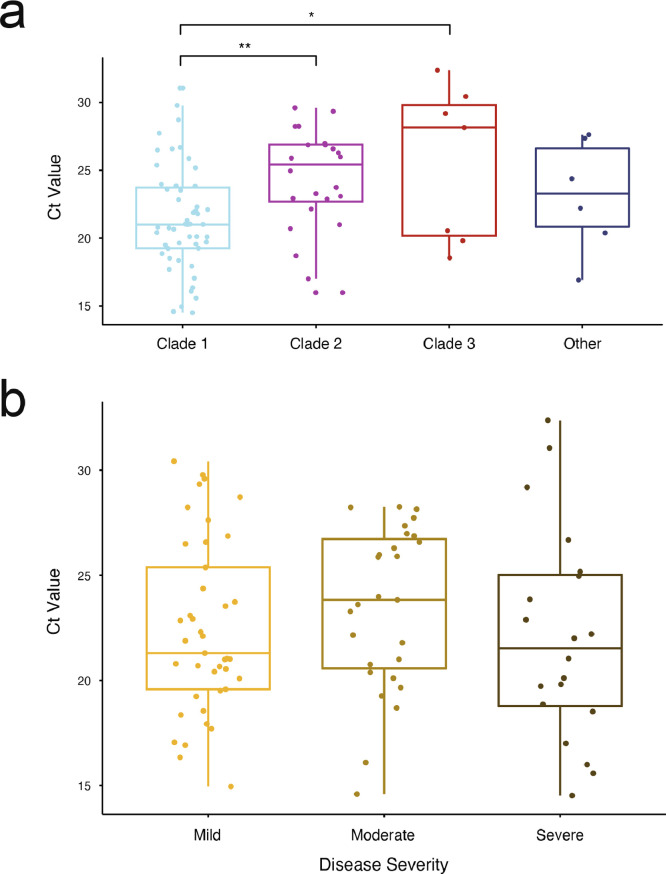
Findings: The 88 SARS-CoV-2 genome sequences in our study separated into three distinct phylogenetic clades. Clades 1 and 3 were most closely related to viral sequences from New York and Washington state, respectively, with relatively broad distributions across the US. Clade 2 was primarily found in the Chicago area with limited distribution elsewhere. At the time of diagnosis, patients infected with Clade 1 viruses had significantly higher average viral loads in their upper airways relative to patients infected with Clade 2 viruses, independent of disease severity.
Interpretation: These results show that multiple variants of SARS-CoV-2 were circulating in the Chicago area in mid-March 2020 that differed in their relative viral loads in patient upper airways. These data suggest that differences in virus genotype can impact viral load and may influence viral spread.
Funding: Dixon Family Translational Research Award, Northwestern University Clinical and Translational Sciences Institute (NUCATS), National Institute of Allergy and Infectious Diseases (NIAID), Lurie Comprehensive Cancer Center, Northwestern University Emerging and Re-emerging Pathogens Program.
Keywords: COVID-19; Phylogenetics; SARS-CoV-2; Viral genotype; Viral load; Whole genome sequencing.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interests The authors declare no competing financial interests.
Figures
Update of
-
A Unique Clade of SARS-CoV-2 Viruses is Associated with Lower Viral Loads in Patient Upper Airways.medRxiv [Preprint]. 2020 Jun 21:2020.05.19.20107144. doi: 10.1101/2020.05.19.20107144. medRxiv. 2020. Update in: EBioMedicine. 2020 Dec;62:103112. doi: 10.1016/j.ebiom.2020.103112. PMID: 32511558 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
