

Pathophysiological mechanisms of liver injury in COVID-19
- PMID: 33190346
- PMCID: PMC7753756
- DOI: 10.1111/liv.14730
Pathophysiological mechanisms of liver injury in COVID-19
Abstract
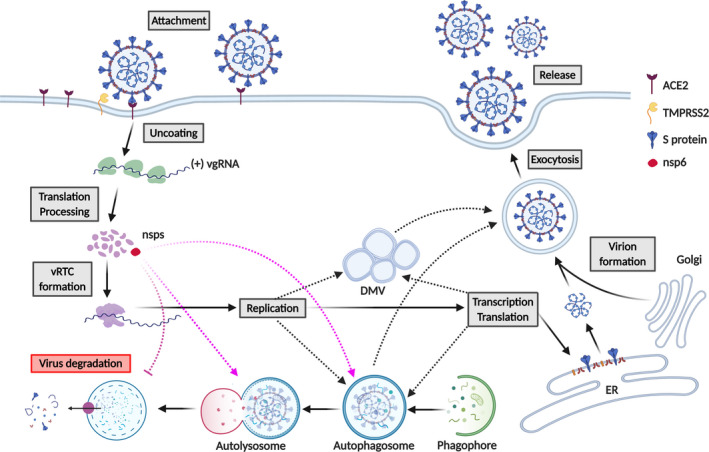
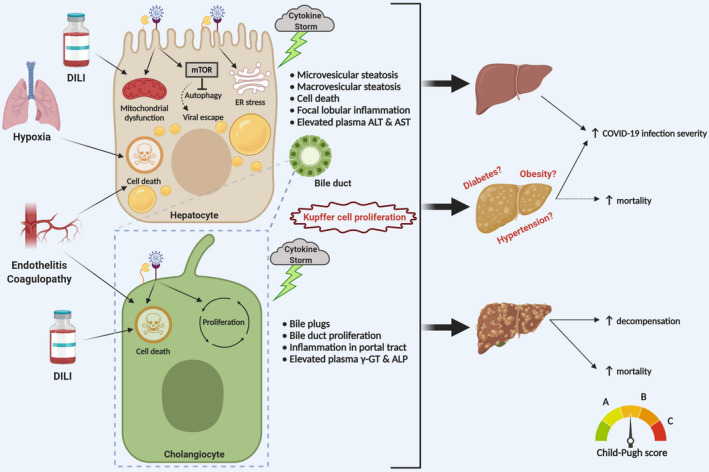
The recent outbreak of coronavirus disease 2019 (COVID-19), caused by the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) has resulted in a world-wide pandemic. Disseminated lung injury with the development of acute respiratory distress syndrome (ARDS) is the main cause of mortality in COVID-19. Although liver failure does not seem to occur in the absence of pre-existing liver disease, hepatic involvement in COVID-19 may correlate with overall disease severity and serve as a prognostic factor for the development of ARDS. The spectrum of liver injury in COVID-19 may range from direct infection by SARS-CoV-2, indirect involvement by systemic inflammation, hypoxic changes, iatrogenic causes such as drugs and ventilation to exacerbation of underlying liver disease. This concise review discusses the potential pathophysiological mechanisms for SARS-CoV-2 hepatic tropism as well as acute and possibly long-term liver injury in COVID-19.
© 2020 The Authors. Liver International published by John Wiley & Sons Ltd.
Conflict of interest statement
The Medical Universities of Graz and Vienna have filed patents for the medical use of
Figures
References
-
- John Hopkins University and Medicine . COVID‐19 Map ‐ Johns Hopkins Coronavirus Resource Center. John Hopkins Coronavirus Resource Center 1 https://coronavirus.jhu.edu/map.html (2020)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
