

Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers
- PMID: 33190502
- PMCID: PMC7775882
- DOI: 10.1021/acs.jproteome.0c00675
Optimizing Accuracy and Depth of Protein Quantification in Experiments Using Isobaric Carriers
Abstract
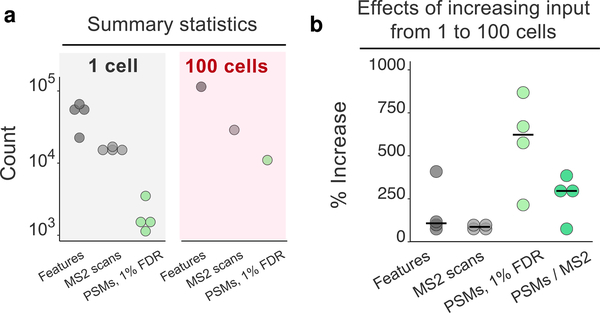
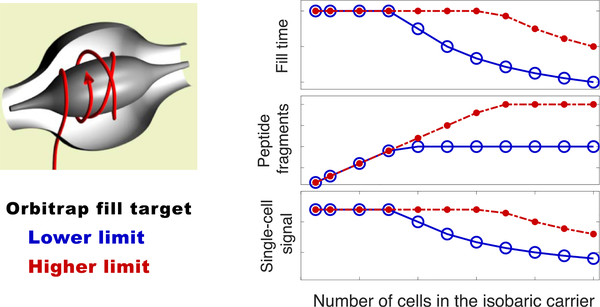
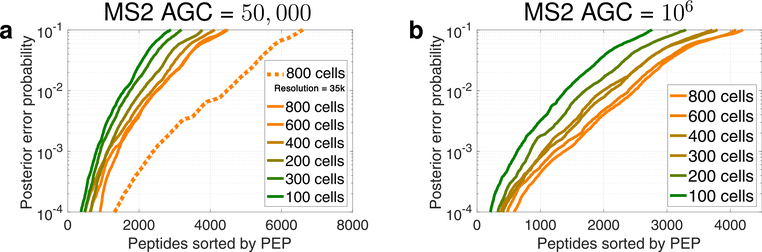
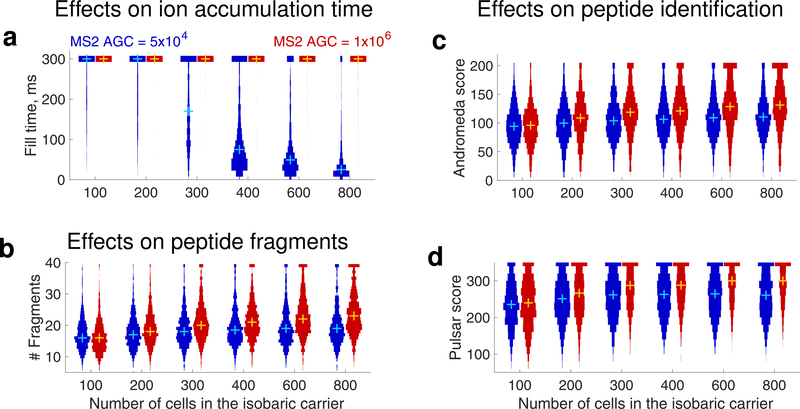
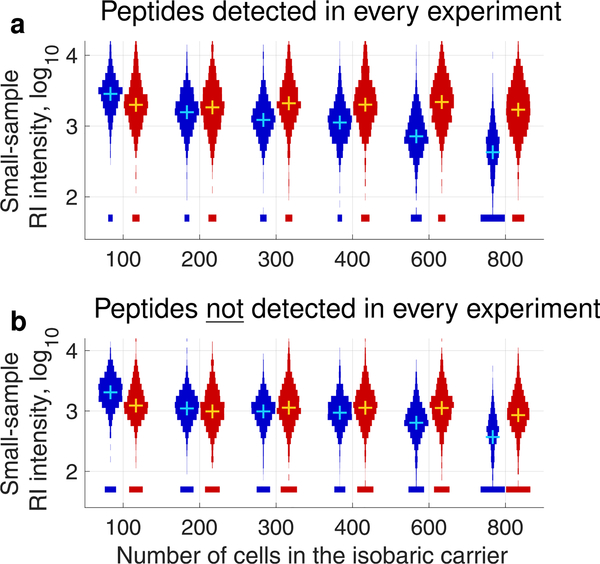
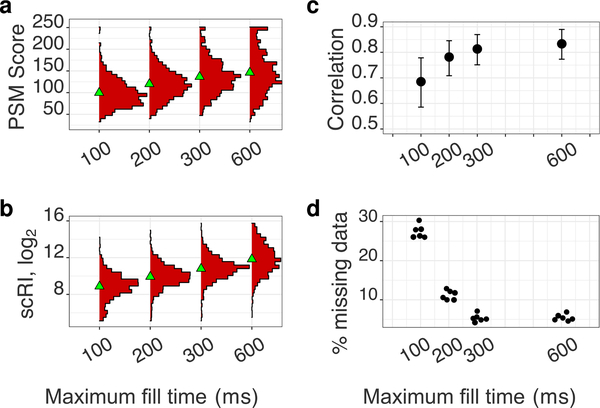
The isobaric carrier approach, which combines small isobarically labeled samples with a larger isobarically labeled carrier sample, finds diverse applications in ultrasensitive mass spectrometry analysis of very small samples, such as single cells. To enhance the growing use of isobaric carriers, we characterized the trade-offs of using isobaric carriers in controlled experiments with complex human proteomes. The data indicate that isobaric carriers directly enhance peptide sequence identification without simultaneously increasing the number of protein copies sampled from small samples. The results also indicate strategies for optimizing the amount of isobaric carrier and analytical parameters, such as ion accumulation time, for different priorities such as improved quantification or an increased number of identified proteins. Balancing these trade-offs enables adapting isobaric carrier experiments to different applications, such as quantifying proteins from limited biopsies or organoids, building single-cell atlases, or modeling protein networks in single cells. In all cases, the reliability of protein quantification should be estimated and incorporated in all subsequent analyses. We expect that these guidelines will aid in explicit incorporation of the characterized trade-offs in experimental designs and transparent error propagation in data analysis.
Keywords: benchmarking; data reliability; isobaric carrier; optimizing mass spectrometry analysis; quantification accuracy; single-cell proteomics.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
