

Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature
- PMID: 33190635
- PMCID: PMC7667763
- DOI: 10.1186/s13395-020-00243-4
Ryanodine receptor 1-related disorders: an historical perspective and proposal for a unified nomenclature
Abstract
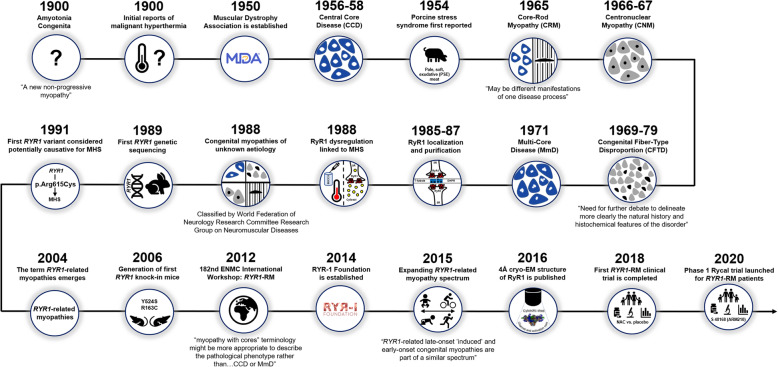
The RYR1 gene, which encodes the sarcoplasmic reticulum calcium release channel or type 1 ryanodine receptor (RyR1) of skeletal muscle, was sequenced in 1988 and RYR1 variations that impair calcium homeostasis and increase susceptibility to malignant hyperthermia were first identified in 1991. Since then, RYR1-related myopathies (RYR1-RM) have been described as rare, histopathologically and clinically heterogeneous, and slowly progressive neuromuscular disorders. RYR1 variants can lead to dysfunctional RyR1-mediated calcium release, malignant hyperthermia susceptibility, elevated oxidative stress, deleterious post-translational modifications, and decreased RyR1 expression. RYR1-RM-affected individuals can present with delayed motor milestones, contractures, scoliosis, ophthalmoplegia, and respiratory insufficiency.Historically, RYR1-RM-affected individuals were diagnosed based on morphologic features observed in muscle biopsies including central cores, cores and rods, central nuclei, fiber type disproportion, and multi-minicores. However, these histopathologic features are not always specific to RYR1-RM and often change over time. As additional phenotypes were associated with RYR1 variations (including King-Denborough syndrome, exercise-induced rhabdomyolysis, lethal multiple pterygium syndrome, adult-onset distal myopathy, atypical periodic paralysis with or without myalgia, mild calf-predominant myopathy, and dusty core disease) the overlap among diagnostic categories is ever increasing. With the continuing emergence of new clinical subtypes along the RYR1 disease spectrum and reports of adult-onset phenotypes, nuanced nomenclatures have been reported (RYR1- [related, related congenital, congenital] myopathies). In this narrative review, we provide historical highlights of RYR1 research, accounts of the main diagnostic disease subtypes and propose RYR1-related disorders (RYR1-RD) as a unified nomenclature to describe this complex and evolving disease spectrum.
Keywords: Clinical neurology; History; Ion channel defects; Myopathy; Neuromuscular disease; Skeletal muscle.
Conflict of interest statement
Dr. Lawal has received support from the RYR-1 Foundation.
Dr. Todd has received support from the RYR-1 Foundation.
Dr. Witherspoon has received support from the RYR-1 Foundation.
Dr. Dirksen is a member of scientific advisory board of the RYR-1 Foundation. Dr. Dirksen has received support from the RYR-1 Foundation, Muscular Dystrophy Association, and the NIH (AR053349).
Dr. Bönnemann is a member of scientific advisory board of the RYR-1 Foundation. Dr. Bönnemann has received funding from Cure CMD.
Dr. Dowling is a member of scientific advisory board of the RYR-1 Foundation and Denature, and a member of the scientific council of the Muscular Dystrophy Association. Dr. Dowling has received support from the RYR-1 Foundation and Muscular Dystrophy Association.
Dr. Hamilton is a member of scientific advisory board of the RYR-1 Foundation. Dr. Hamilton has received the following support from the NIH: R01AR072602, R01AR072475, and R01AR053349. Dr. Hamilton has also received a grant from the Muscular Dystrophy Association and a gift from the McNair Foundation.
Dr. Meilleur has received support from the RYR-1 Foundation.
Figures


References
-
- Dubowitz V. The “new” myopathies. Neuropediatrics. 1969;1(02):137–148. doi: 10.1055/s-0028-1091869. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
