

Genotype-Phenotype Associations in 72 Adults with Suspected ALPL-Associated Hypophosphatasia
- PMID: 33191482
- PMCID: PMC7881968
- DOI: 10.1007/s00223-020-00771-7
Genotype-Phenotype Associations in 72 Adults with Suspected ALPL-Associated Hypophosphatasia
Abstract
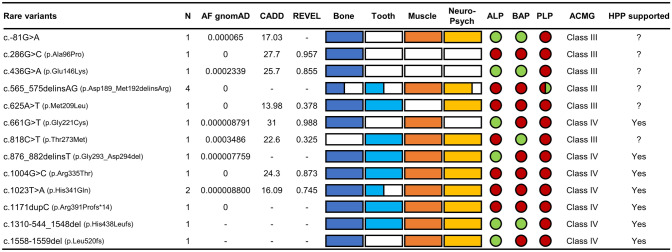
Hypophosphatasia (HPP) is a rare inborn error of metabolism due to a decreased activity of tissue nonspecific alkaline phosphatase (TNSALP). As the onset and severity of HPP are heterogenous, it can be challenging to determine the pathogenicity of detected rare ALPL variants in symptomatic patients. We aimed to characterize patients with rare ALPL variants to propose which patients can be diagnosed with adult HPP. We included 72 patients with (1) clinical symptoms of adult HPP or positive family history and (2) low TNSALP activity and/or high pyridoxal 5'-phosphate (PLP) levels, who underwent ALPL gene sequencing. The patients were analyzed and divided into three groups depending on ALPL variant pathogenicity according to the classification of the American College of Medical Genetics and Genomics (ACMG). Reported pathogenic (n = 34 patients), rare (n = 17) and common (n = 21) ALPL variants only were found. Muscular complaints were the most frequent symptoms (> 80%), followed by bone affection (> 50%). Tooth involvement was significantly more common in patients with pathogenic or rare ALPL variants. Seven rare variants could be classified as likely pathogenic (ACMG class 4) of which five have not yet been described. Inconclusive genetic findings and less specific symptoms make diagnosis difficult in cases where adult HPP is not obvious. As not every pathogenic or rare ALPL variant leads to a manifestation of HPP, only patients with bone complications and at least one additional complication concerning teeth, muscle, central nervous and mental system, repeated low TNSALP activity and high PLP levels should be diagnosed as adult HPP if rare ALPL gene variants of ACMG class 4 or higher support the diagnosis.
Keywords: ALP; Alkaline phosphatase; HPP; PLP; Pyridoxal 5′-phosphate; TNSALP.
Conflict of interest statement
Nico Maximilian Jandl, Tobias Schmidt, Tim Rolvien, Julian Stürznickel, Konstantin Chrysostomou, Emil von Vopelius, Alexander E. Volk, Thorsten Schinke, Christian Kubisch and Michael Amling declare that they have no conflict of interest. Florian Barvencik received speaker and consultant fees from Alexion.
Figures



References
-
- Whyte MP. Hypophosphatasia-aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12:233–246. - PubMed
-
- Mornet E. Hypophosphatasia. Metabol. 2018;82:142–155. - PubMed
-
- Khan AA, Josse R, Kannu P, Villeneuve J, Paul T, Van Uum S, Greenberg CR. Hypophosphatasia: Canadian update on diagnosis and management. Osteoporos Int. 2019;30:1713–1722. - PubMed
-
- Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75:439–445. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
