

Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic
- PMID: 33193132
- PMCID: PMC7644918
- DOI: 10.3389/fmicb.2020.550674
Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic
Abstract
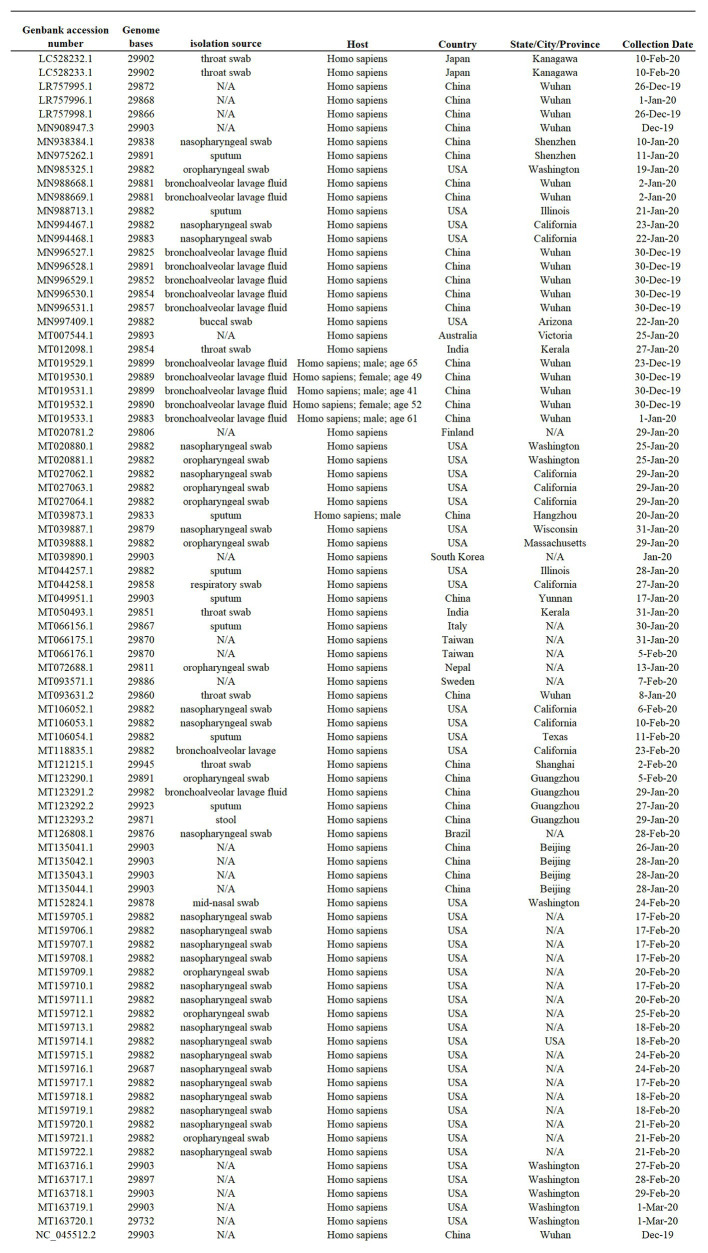
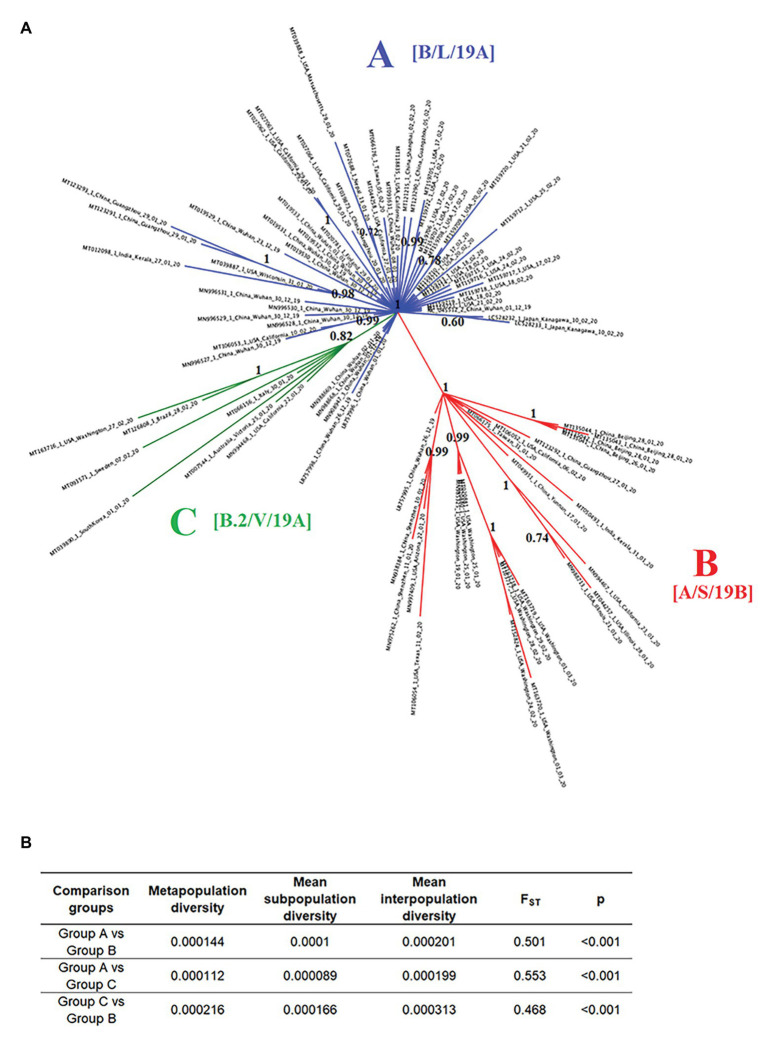
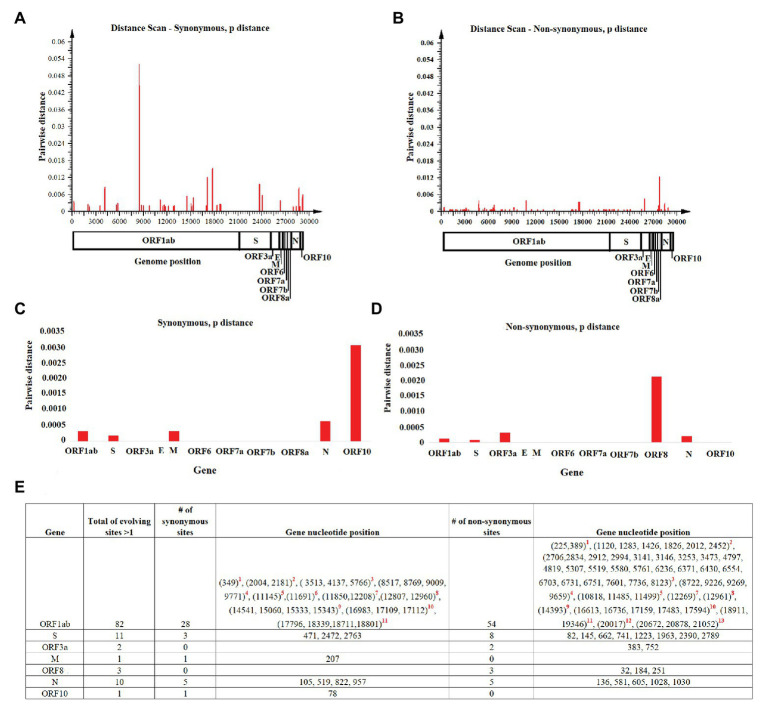
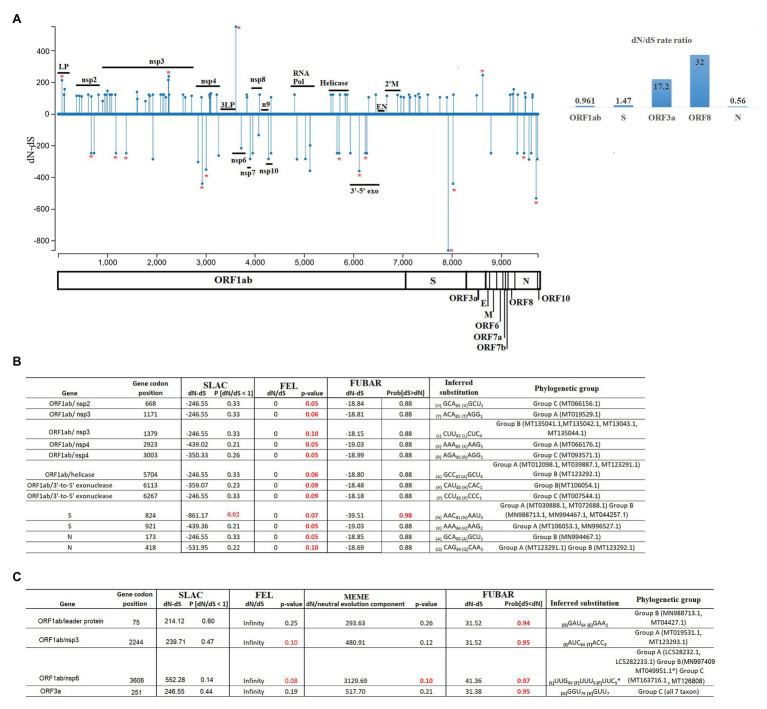
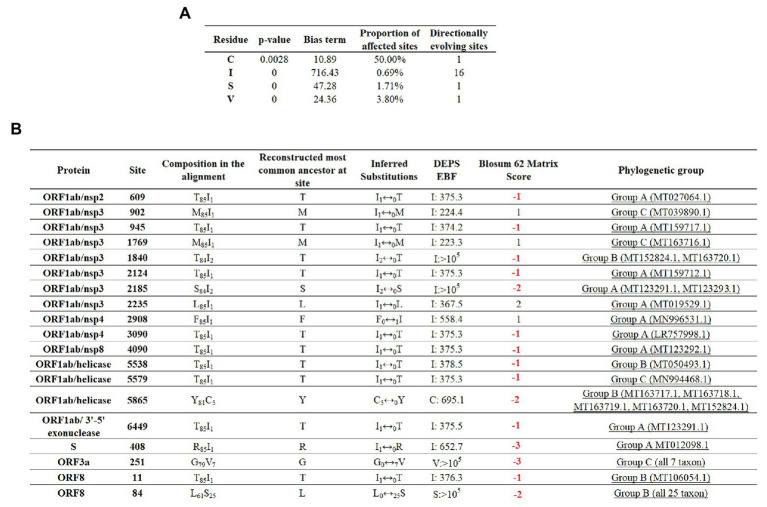
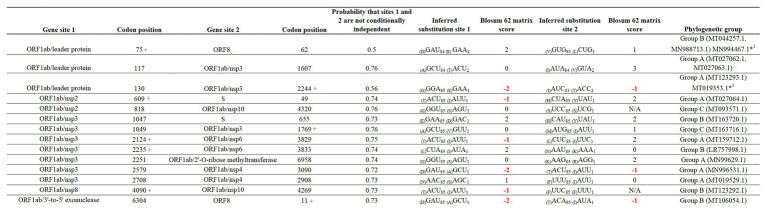
In this study, we analyzed full-length SARS-CoV-2 genomes from multiple countries to determine early trends in the evolutionary dynamics of the novel COVID-19 pandemic. Results indicated SARS-CoV-2 evolved early into at least three phylogenetic groups, characterized by positive selection at specific residues of the accessory proteins ORF3a and ORF8. Also, we are reporting potential relevant sites under positive selection at specific sites of non-structural proteins nsp6 and helicase. Our analysis of co-evolution showed evidence of epistatic interactions among sites in the genome that may be important in the generation of variants adapted to humans. These observations might impact not only public health but also suggest that more studies are needed to understand the genetic mechanisms that may affect the development of therapeutic and preventive tools, like antivirals and vaccines. Collectively, our results highlight the identification of ongoing selection even in a scenario of conserved sequences collected over the first 3 months of this pandemic.
Keywords: COVID-19; SARS-CoV2; epistasis; evolution; positive selection.
Copyright © 2020 Velazquez-Salinas, Zarate, Eberl, Gladue, Novella and Borca.
Figures
References
-
- Alm E., Broberg E. K., Connor T., Hodcroft E. B., Komissarov A. B., Maurer-Stroh S., et al. . (2020). Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European region, January to June 2020. Euro Surveill. 25:2001410. 10.2807/1560-7917.ES.2020.25.32.2001410, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous