

PI3K γ Regulatory Protein p84 Determines Mast Cell Sensitivity to Ras Inhibition-Moving Towards Cell Specific PI3K Targeting?
- PMID: 33193405
- PMCID: PMC7655736
- DOI: 10.3389/fimmu.2020.585070
PI3K γ Regulatory Protein p84 Determines Mast Cell Sensitivity to Ras Inhibition-Moving Towards Cell Specific PI3K Targeting?
Abstract
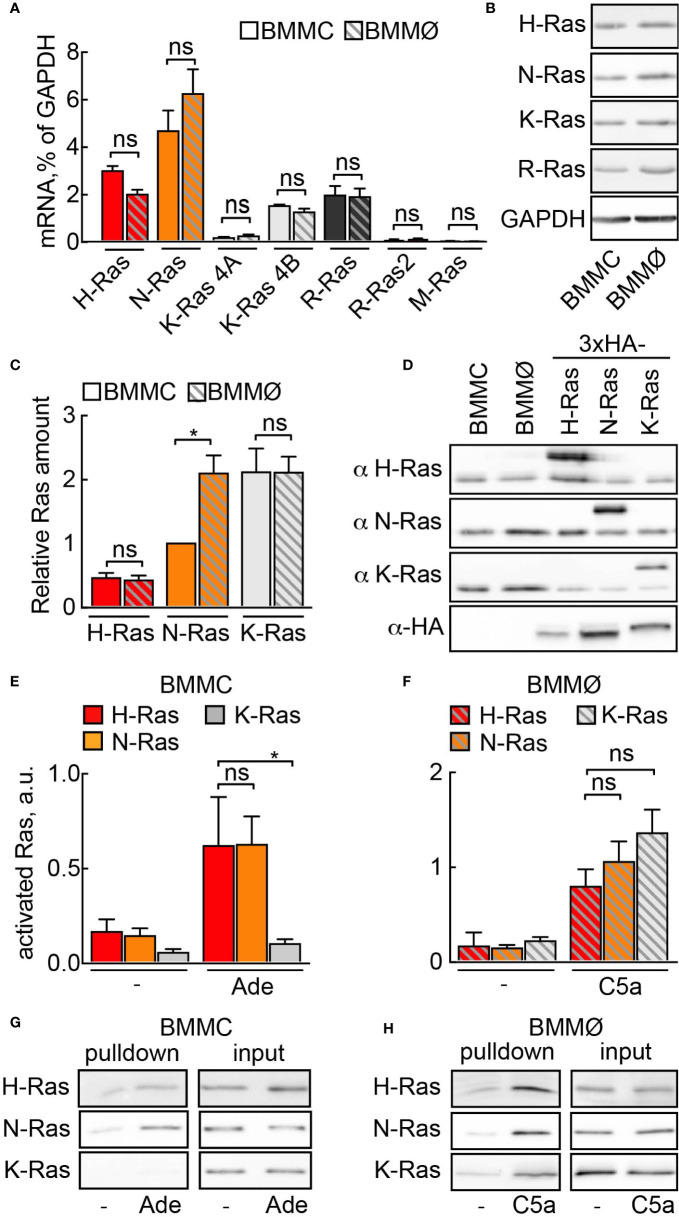
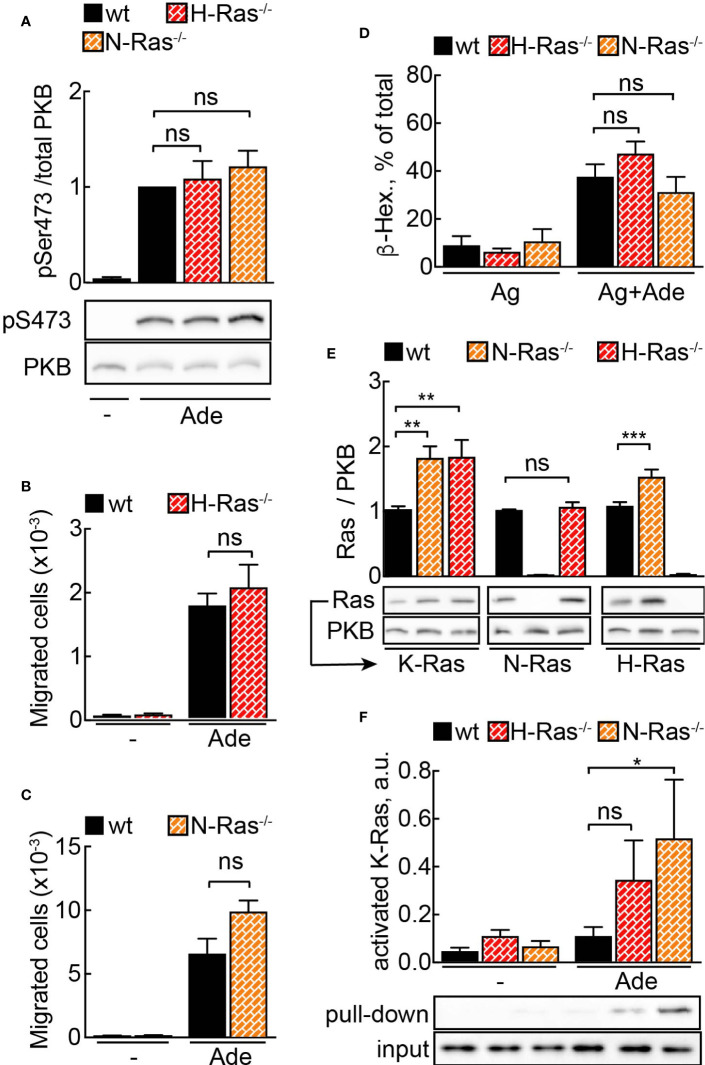
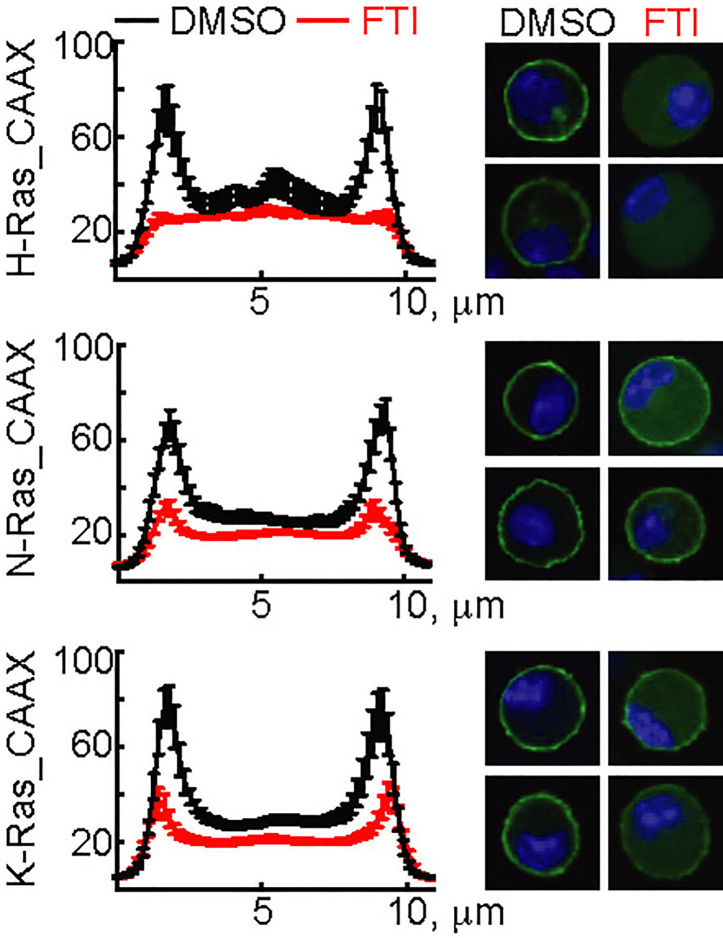
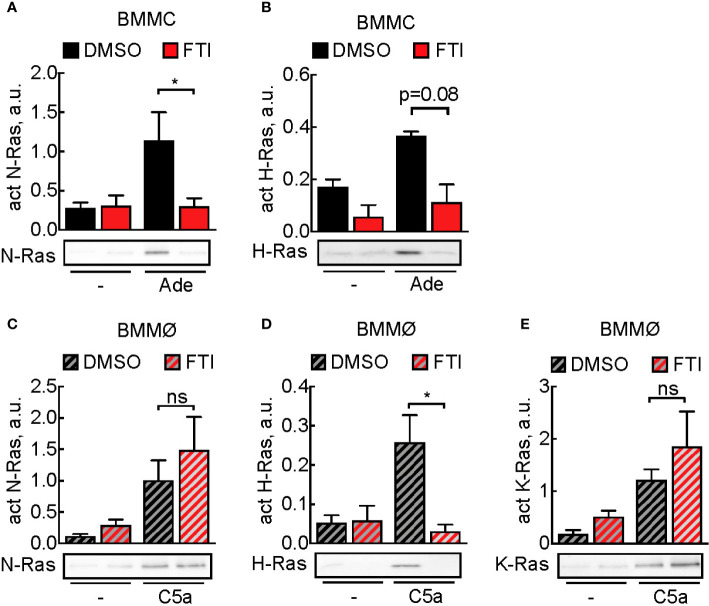
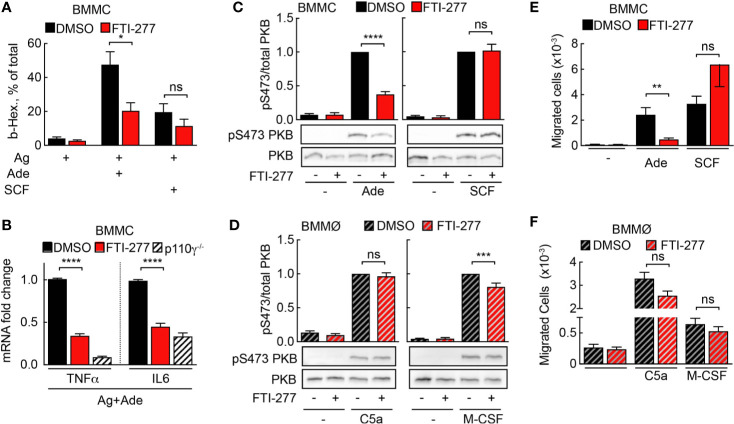
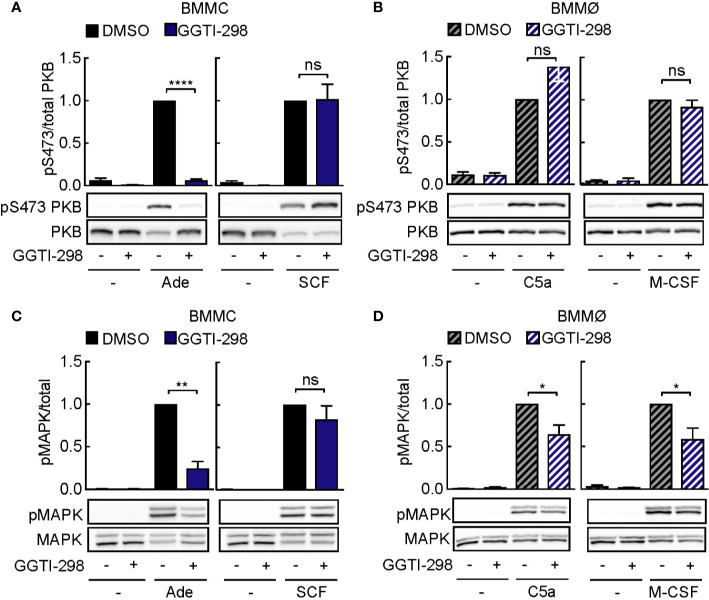
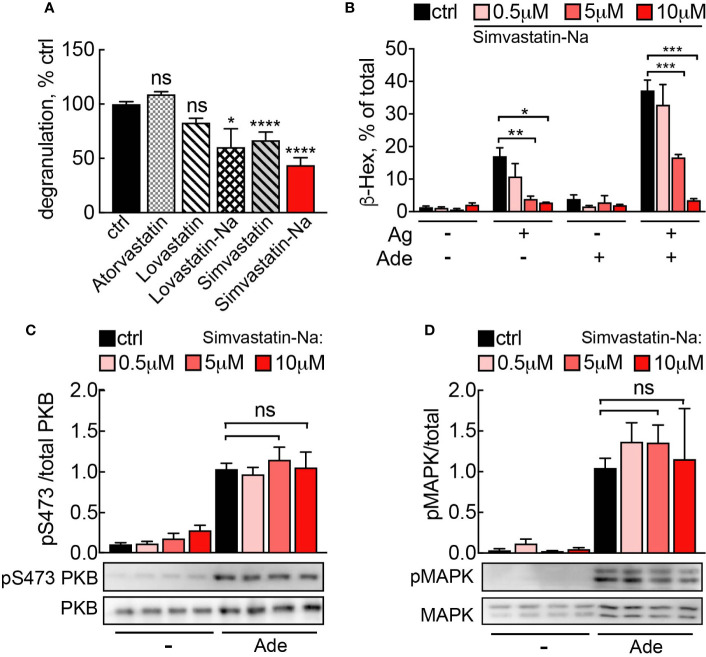
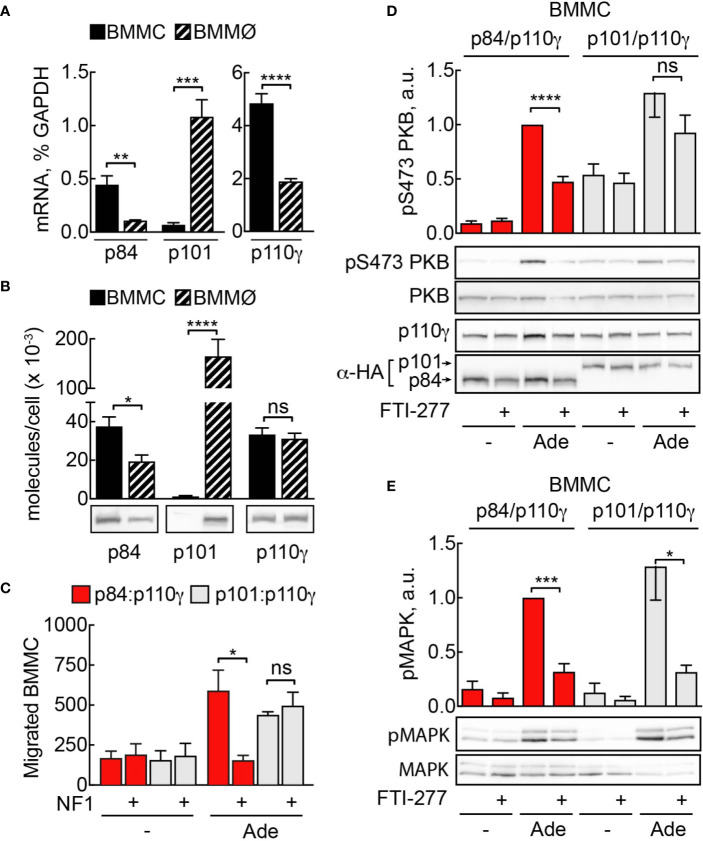
Mast cells are the major effector cells in immunoglobulin E (IgE)-mediated allergy. The high affinity IgE receptor FcεRI, as well as G protein-coupled receptors (GPCRs) on the mast cell surface signals to phosphoinositide 3-kinase γ (PI3Kγ) to initiate degranulation, cytokine release, and chemotaxis. PI3Kγ is therefore considered as a target for treatment of allergic disorders. However, leukocyte PI3Kγ is key to many functions in innate and adaptive immunity, and attenuation of host defense mechanisms is an expected adverse effect that complicates treatment of chronic illnesses. PI3Kγ operates as a p110γ/p84 or p110γ/p101 complex, where p110γ/p84 requires Ras activation. Here we investigated if modulation of Ras-isoprenylation could target PI3Kγ activity to attenuate PI3Kγ-dependent mast cell responses without impairment of macrophage functions. In murine bone marrow-derived mast cells, GPCR stimulation triggers activation of N-Ras and H-Ras isoforms, which is followed by the phosphorylation of protein kinase B (PKB/Akt) relayed through PI3Kγ. Although K-Ras is normally not activated in Ras wild-type cells, it is able to compensate for genetically deleted N- and H-Ras isoforms. Inhibition of Ras isoprenylation with farnesyltransferase inhibitor FTI-277 leads to a significant reduction of mast cell degranulation, cytokine production, and migration. Complementation experiments expressing PI3Kγ adaptor proteins p84 or p101 demonstrated a differential sensitivity towards Ras-inhibition depending on PI3Kγ complex composition. Mast cell responses are exclusively p84-dependent and were effectively controlled by FTI-277. Similar results were obtained when GTP-Ras was inactivated by overexpression of the GAP-domain of Neurofibromin-1 (NF-1). Unlike mast cells, macrophages express p84 and p101 but are p101-dominated and thus remain functional under treatment with FTI-277. Our work demonstrates that p101 and p84 have distinct physiological roles, and that Ras dependence of PI3Kγ signaling differs between cell types. FTI-277 reduces GPCR-activated PI3Kγ responses in p84-expressing but not p101-containing bone marrow derived cells. However, prenylation inhibitors have pleiotropic effects beyond Ras and non-tolerable side-effects that disfavor further clinical validation. Statins are, however, clinically well-established drugs that have previously been proposed to block mast cell degranulation by interference with protein prenylation. We show here that Simvastatin inhibits mast cell degranulation, but that this does not occur via Ras-PI3Kγ pathway alterations.
Keywords: IgE (Immunoglobulin E); PIK3CG; Ras family proteins; allergy; inflammation; p101; p84; phosphoinositide-3-kinase (PI3K).
Copyright © 2020 Jin, Gogvadze, Xavier, Bohnacker, Voelzmann and Wymann.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous