

BH3 Mimetics for the Treatment of B-Cell Malignancies-Insights and Lessons from the Clinic
- PMID: 33198338
- PMCID: PMC7696913
- DOI: 10.3390/cancers12113353
BH3 Mimetics for the Treatment of B-Cell Malignancies-Insights and Lessons from the Clinic
Abstract
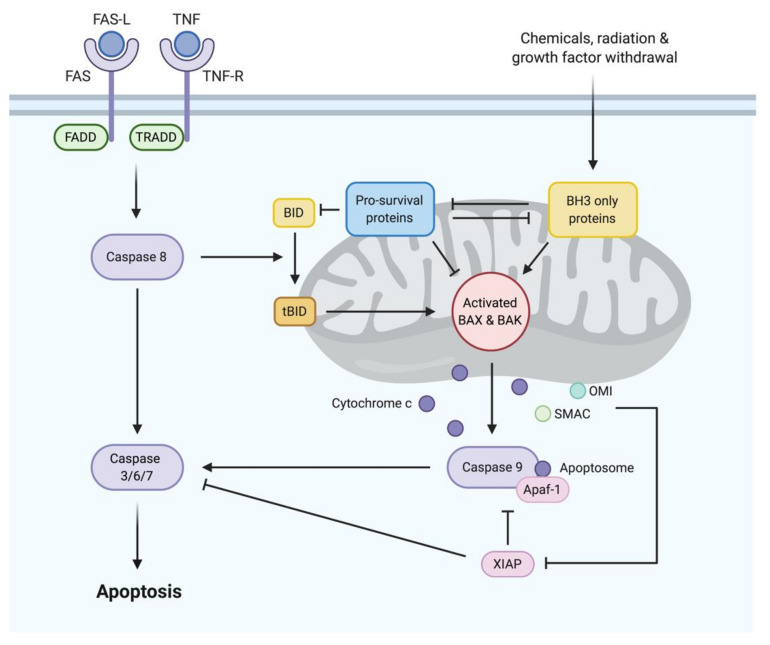
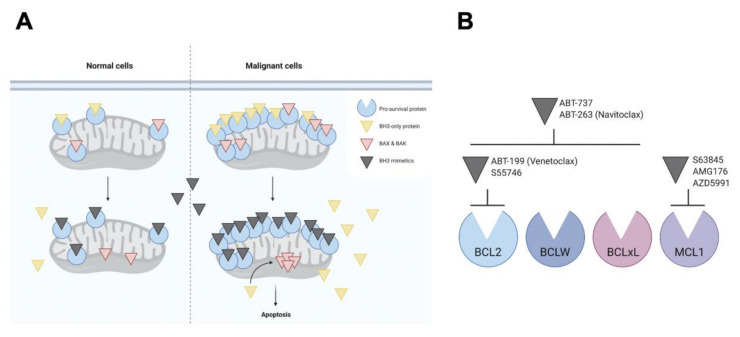
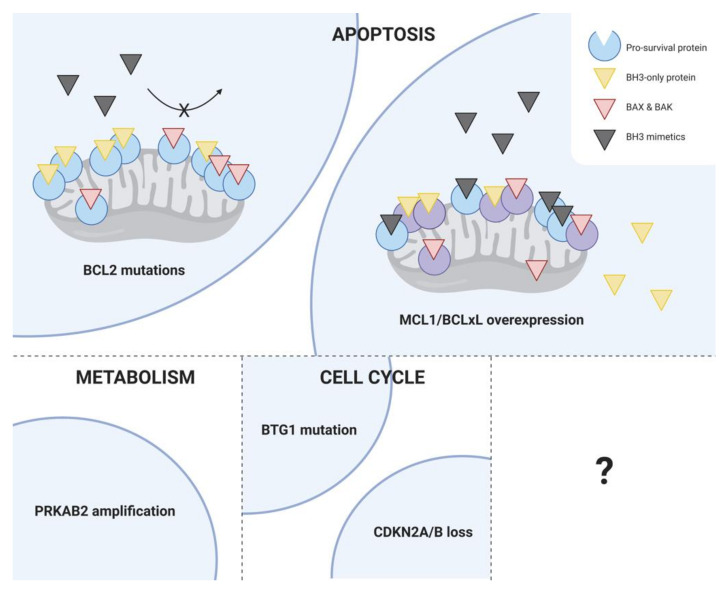
The discovery of the link between defective apoptotic regulation and cancer cell survival engendered the idea of targeting aberrant components of the apoptotic machinery for cancer therapy. The intrinsic pathway of apoptosis is tightly controlled by interactions amongst members of three distinct subgroups of the B-cell lymphoma 2 (BCL2) family of proteins. The pro-survival BCL2 proteins prevent apoptosis by keeping the pro-apoptotic effector proteins BCL2-associated X protein (BAX) and BCL2 homologous antagonist/killer (BAK) in check, while the BH3-only proteins initiate apoptosis by either neutralizing the pro-survival BCL2 proteins or directly activating the pro-apoptotic effector proteins. This tripartite regulatory mechanism is commonly perturbed in B-cell malignancies facilitating cell death evasion. Over the past two decades, structure-based drug discovery has resulted in the development of a series of small molecules that mimic the function of BH3-only proteins called the BH3 mimetics. The most clinically advanced of these is venetoclax, which is a highly selective inhibitor of BCL2 that has transformed the treatment landscape for chronic lymphocytic leukemia (CLL). Other BH3 mimetics, which selectively target myeloid cell leukemia 1 (MCL1) and B-cell lymphoma extra large (BCLxL), are currently under investigation for use in diverse malignancies. Here, we review the current role of BH3 mimetics in the treatment of CLL and other B-cell malignancies and address open questions in this rapidly evolving field.
Keywords: B-cell malignancies; BCL2; BCLxL; BH3 mimetics; MCL1; apoptosis; leukemia; lymphoma; myeloma; venetoclax.
Conflict of interest statement
D.C.S.H. and R.T. are employees of the Walter and Eliza Hall Institute of Medical Research, which receives milestone and royalty payments related to venetoclax. D.C.S.H. has received research funding from Genentech and Servier.
Figures
References
-
- Kroemer G., Galluzzi L., Vandenabeele P., Abrams J., Alnemri E.S., Baehrecke E.H., Blagosklonny M.V., El-Deiry W.S., Golstein P., Green D.R., et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Diff. 2008;16:3–11. doi: 10.1038/cdd.2008.150. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
