

Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life
- PMID: 33199898
- PMCID: PMC7783656
- DOI: 10.1038/s41593-020-00737-w
Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life
Abstract
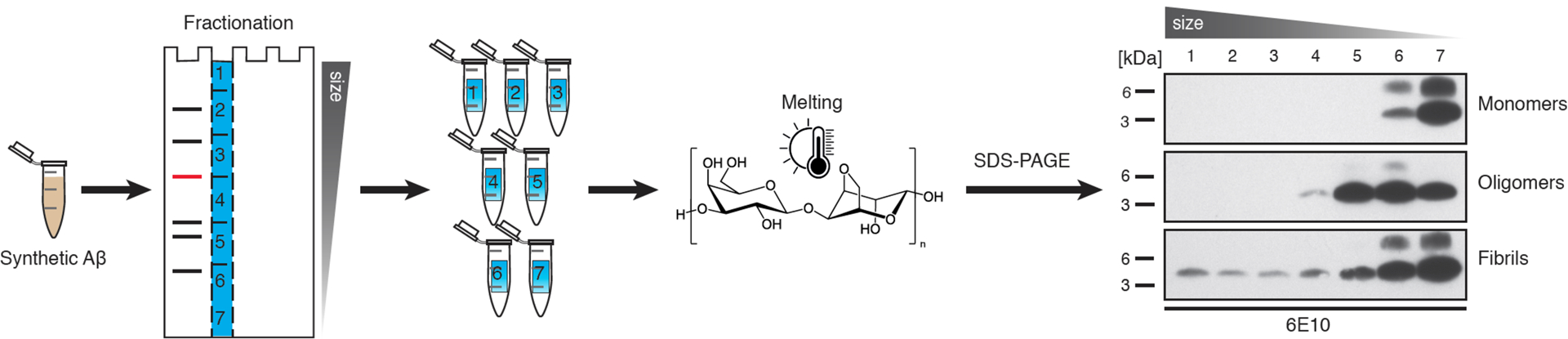
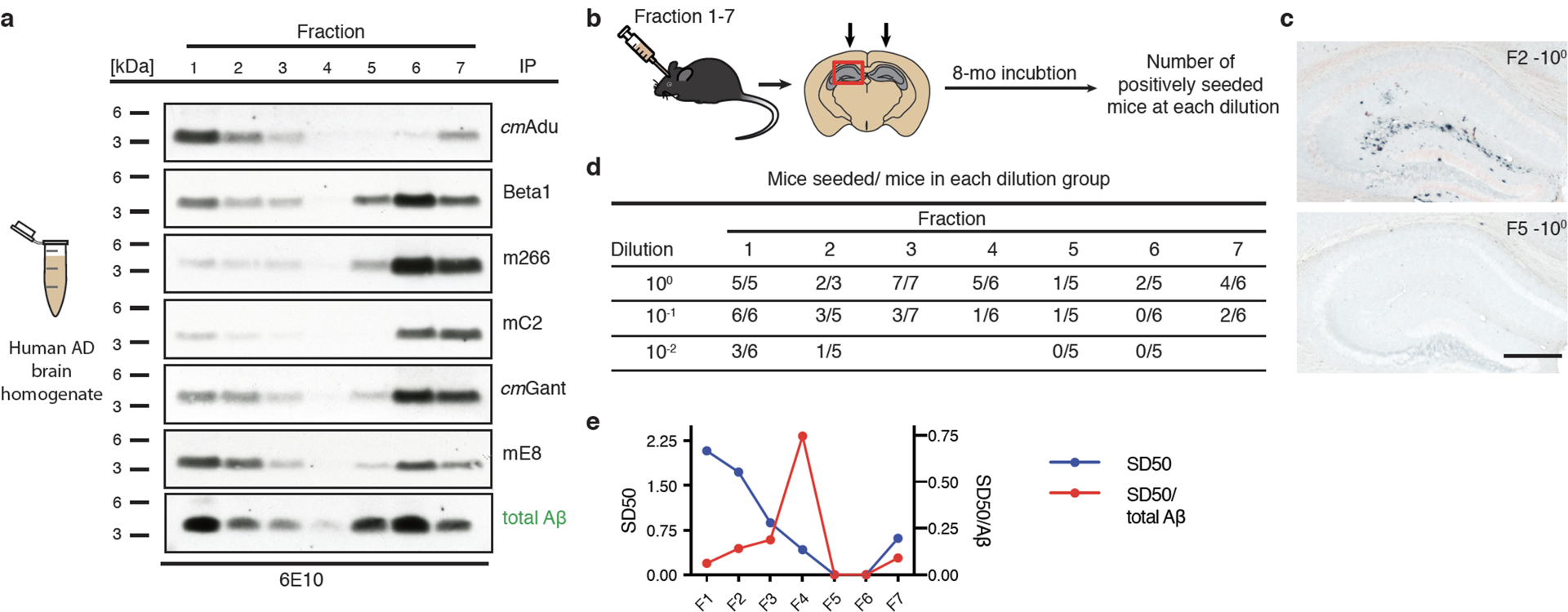
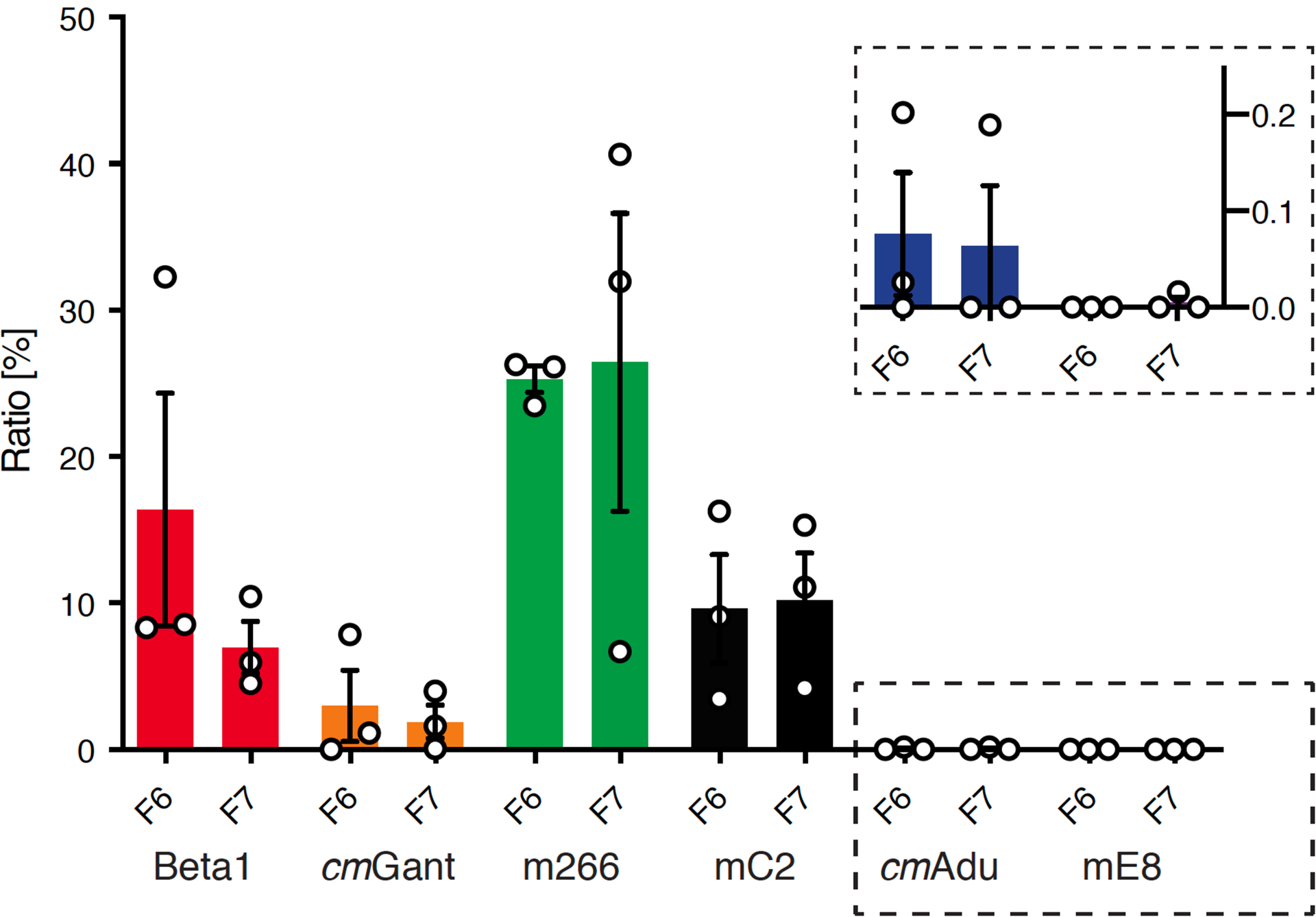
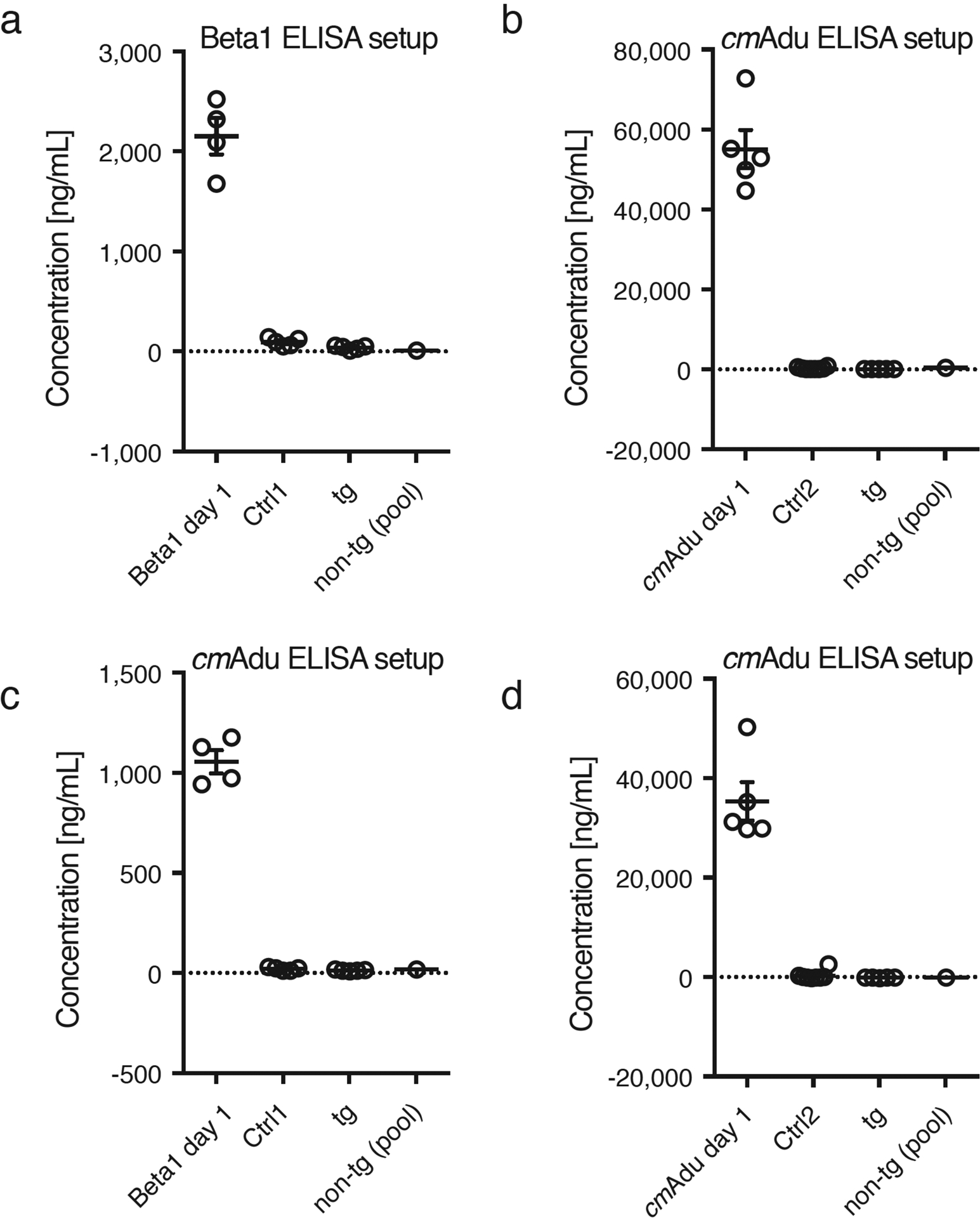
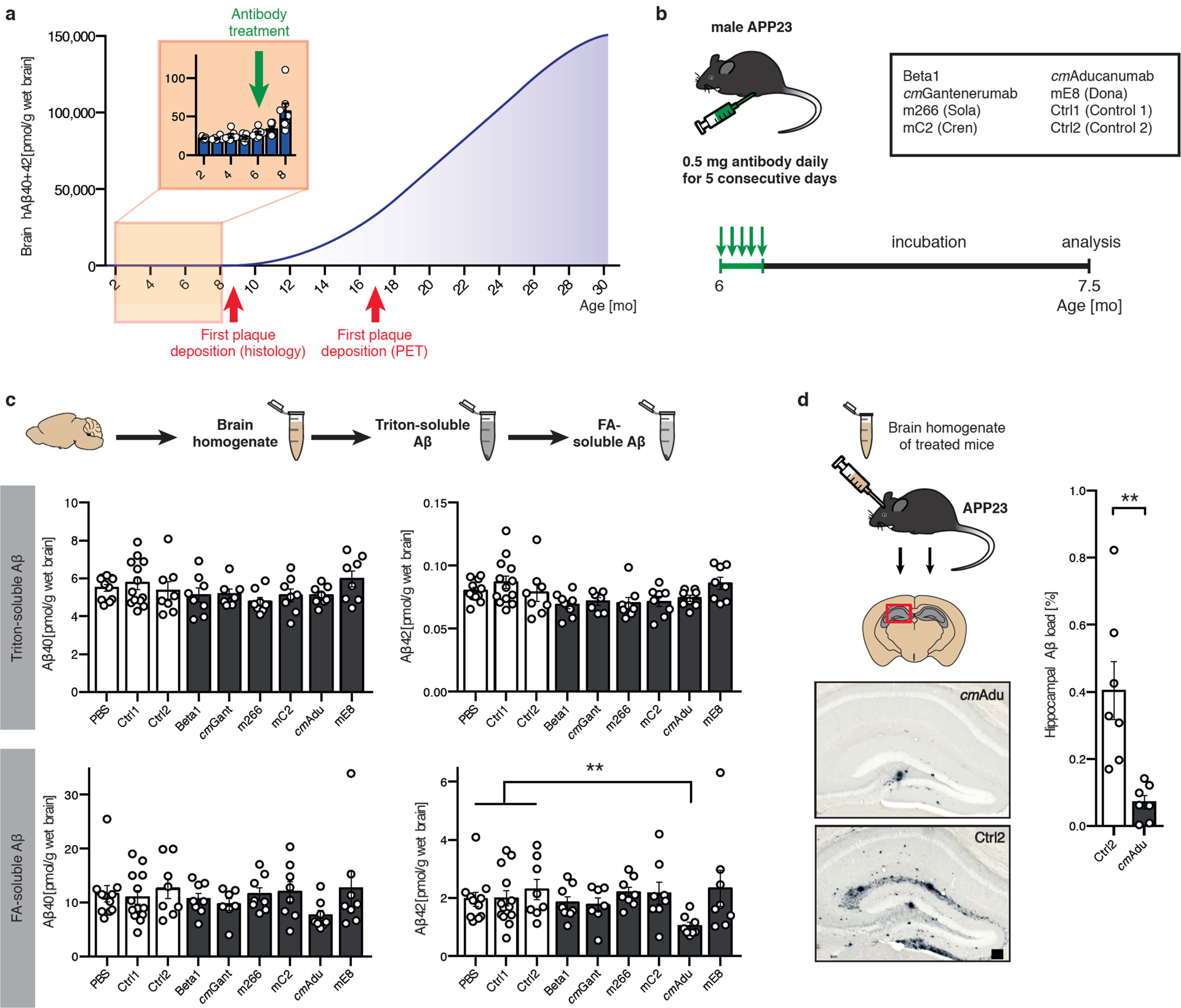
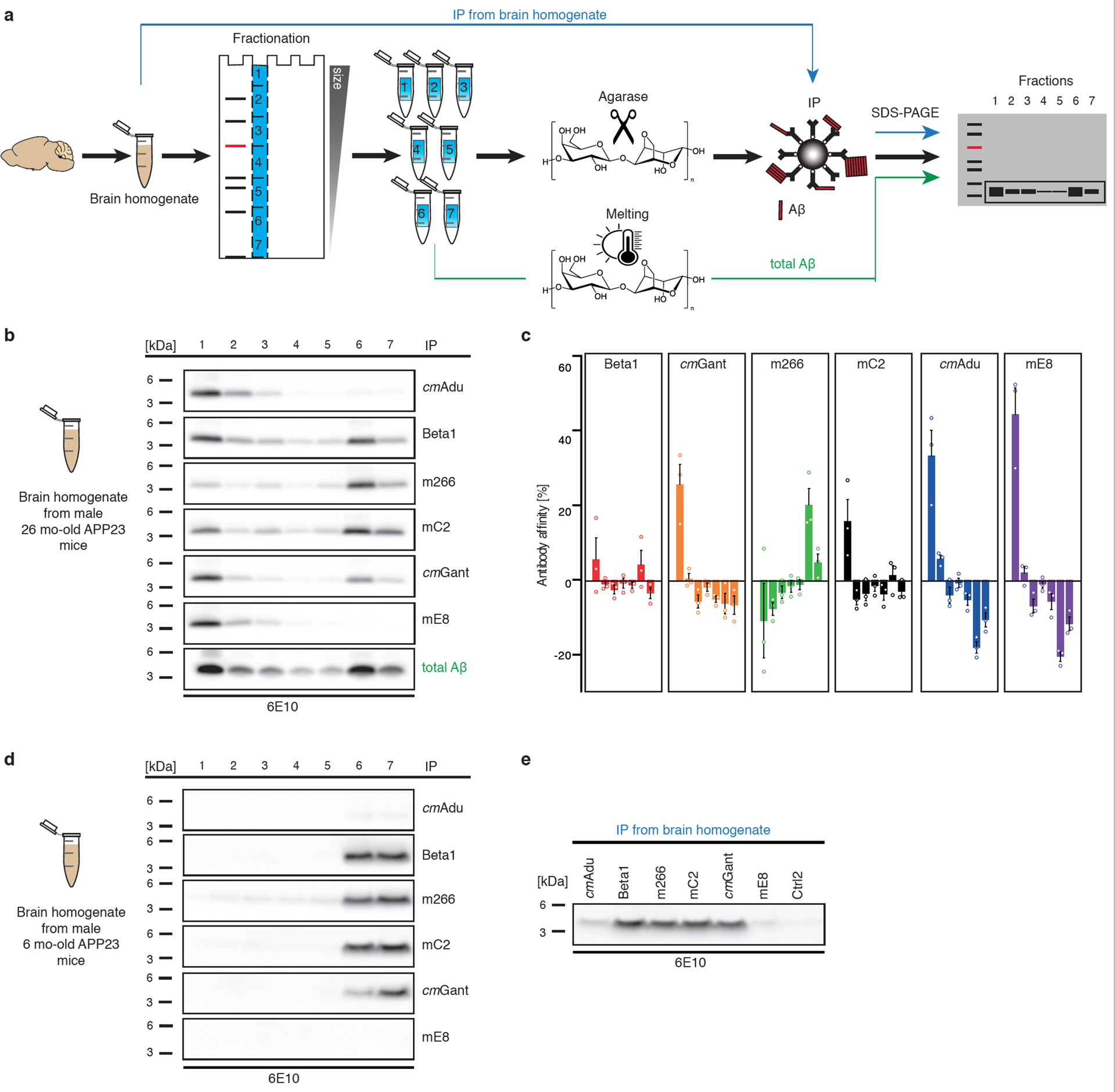
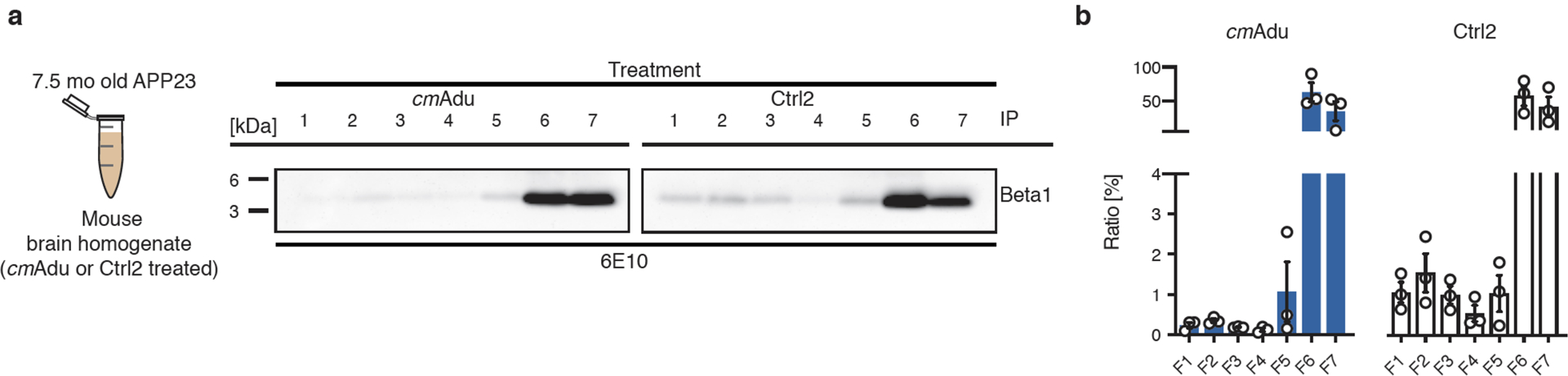
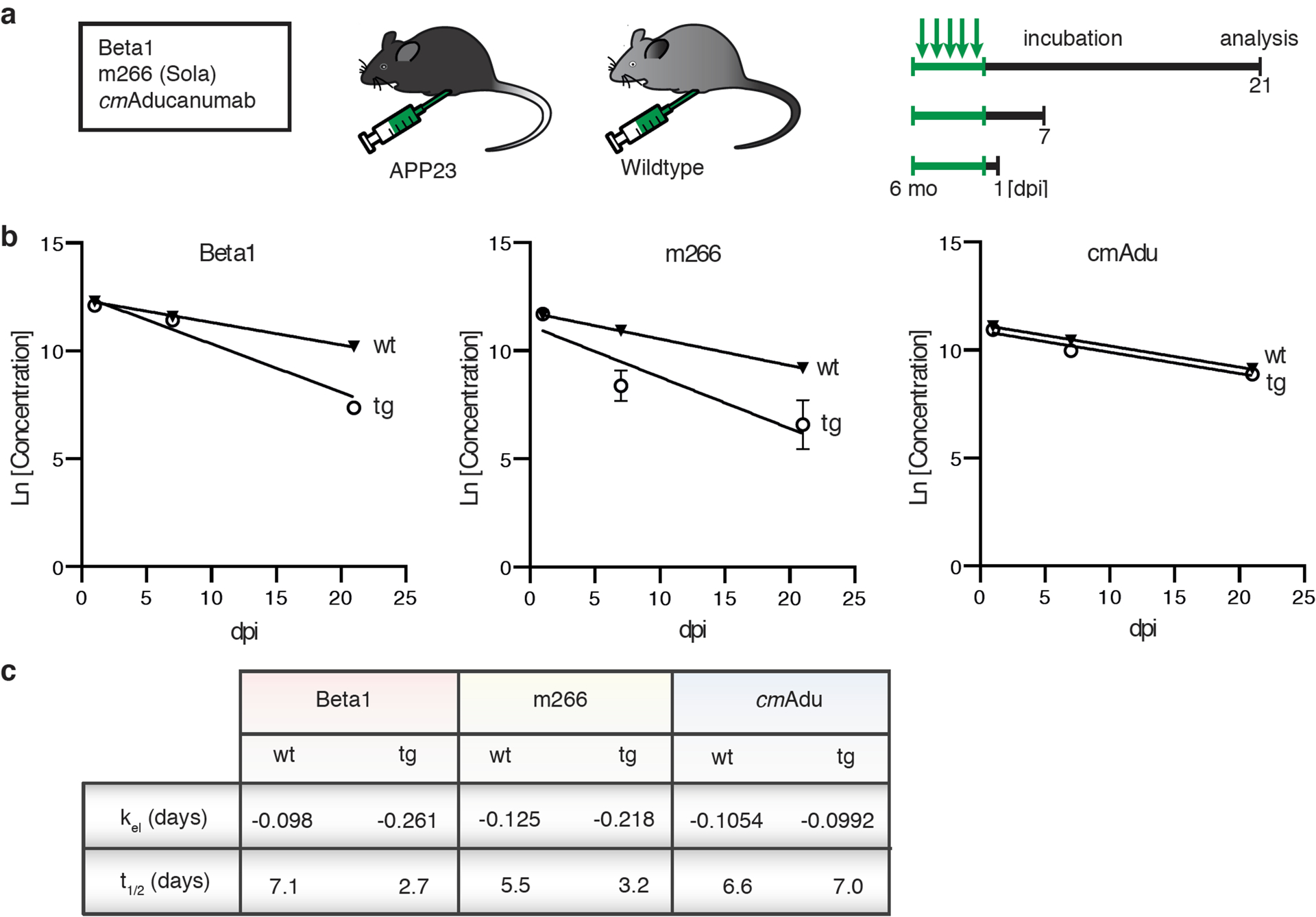
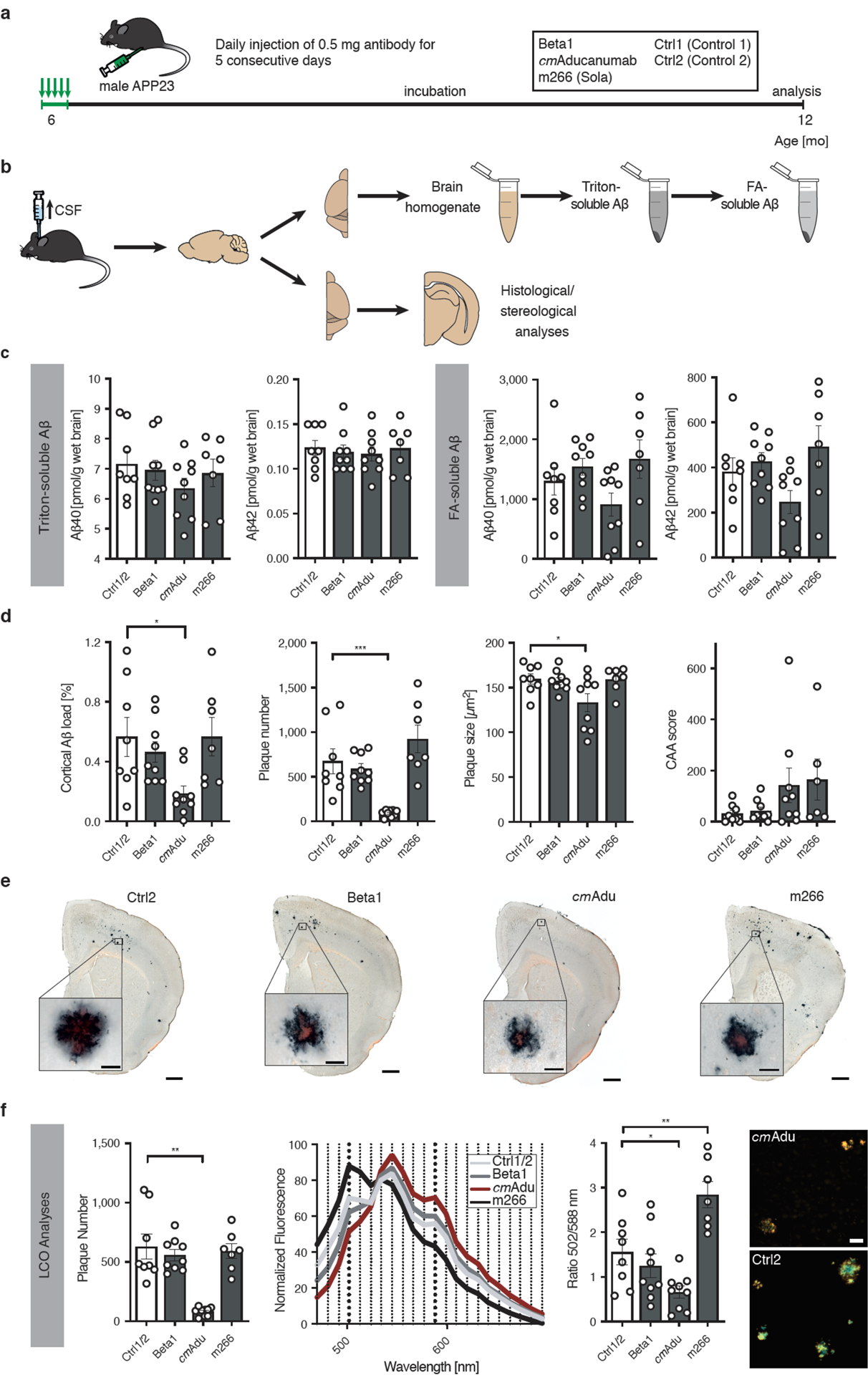
Amyloid-β (Aβ) deposits are a relatively late consequence of Aβ aggregation in Alzheimer's disease. When pathogenic Aβ seeds begin to form, propagate and spread is not known, nor are they biochemically defined. We tested various antibodies for their ability to neutralize Aβ seeds before Aβ deposition becomes detectable in Aβ precursor protein-transgenic mice. We also characterized the different antibody recognition profiles using immunoprecipitation of size-fractionated, native, mouse and human brain-derived Aβ assemblies. At least one antibody, aducanumab, after acute administration at the pre-amyloid stage, led to a significant reduction of Aβ deposition and downstream pathologies 6 months later. This demonstrates that therapeutically targetable pathogenic Aβ seeds already exist during the lag phase of protein aggregation in the brain. Thus, the preclinical phase of Alzheimer's disease-currently defined as Aβ deposition without clinical symptoms-may be a relatively late manifestation of a much earlier pathogenic seed formation and propagation that currently escapes detection in vivo.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS:
S.C., F.K., and J.B.S are current or former employees of Lundbeck; F.Q., P.H.W., and T.B. are current employees and/or shareholders of Biogen; J.-U.R and H.C. are former employees of Probiodrug AG; M.S. is a former employee of Novartis. The other authors have no competing interest.
Figures

References
-
- Panza F, Lozupone M, Seripa D & Imbimbo BP Amyloid-β immunotherapy for Alzheimer disease: Is it now a long shot? Ann. Neurol 85, 303–315 (2019). - PubMed
-
- Villemagne VL, Doré V, Burnham SC, Masters CL & Rowe CC Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol 14, 225–236 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
