

Detection of Pathogenic Variants With Germline Genetic Testing Using Deep Learning vs Standard Methods in Patients With Prostate Cancer and Melanoma
- PMID: 33201204
- PMCID: PMC7672519
- DOI: 10.1001/jama.2020.20457
Detection of Pathogenic Variants With Germline Genetic Testing Using Deep Learning vs Standard Methods in Patients With Prostate Cancer and Melanoma
Abstract
Importance: Less than 10% of patients with cancer have detectable pathogenic germline alterations, which may be partially due to incomplete pathogenic variant detection.
Objective: To evaluate if deep learning approaches identify more germline pathogenic variants in patients with cancer.
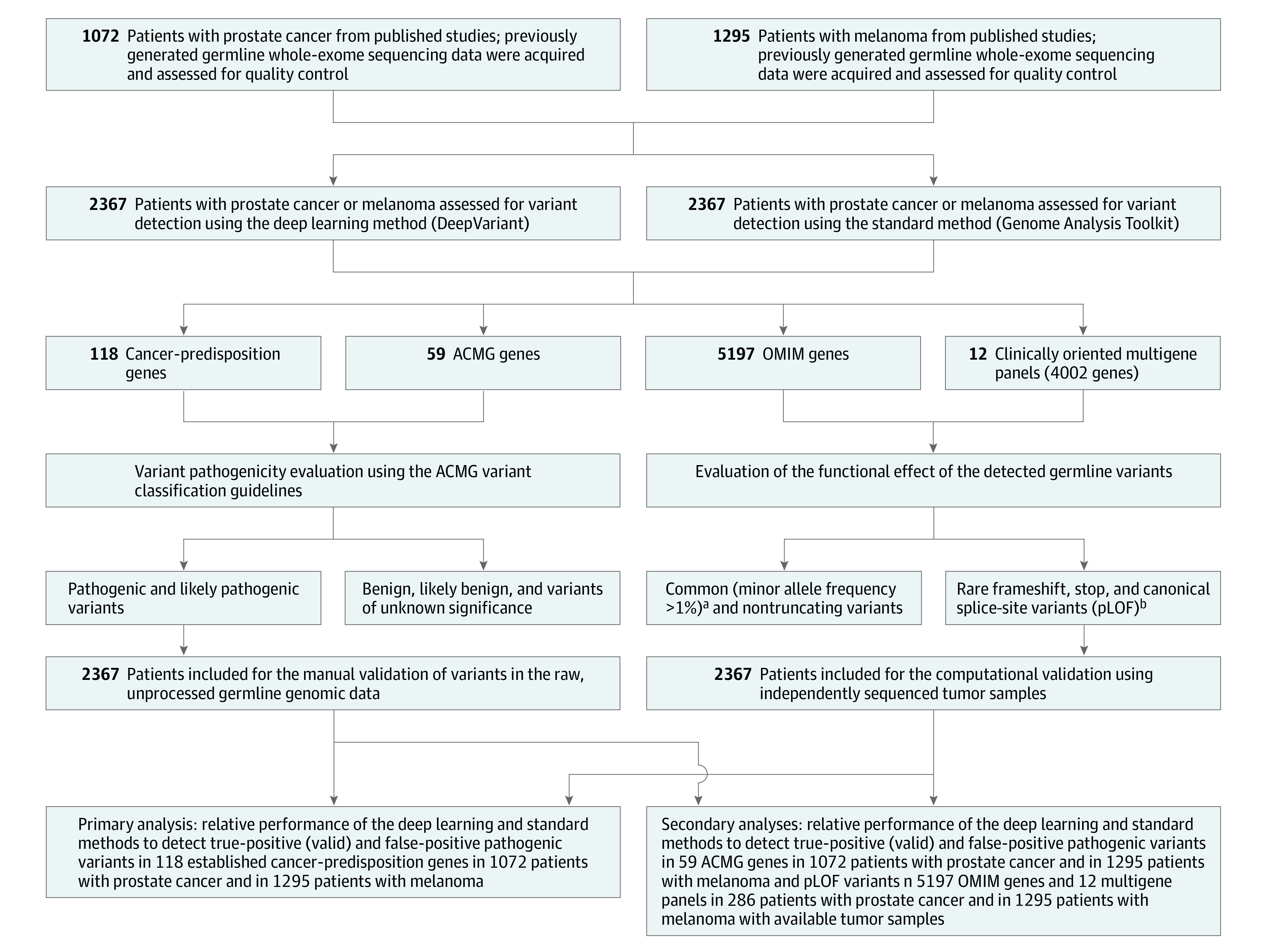
Design, setting, and participants: A cross-sectional study of a standard germline detection method and a deep learning method in 2 convenience cohorts with prostate cancer and melanoma enrolled in the US and Europe between 2010 and 2017. The final date of clinical data collection was December 2017.
Exposures: Germline variant detection using standard or deep learning methods.
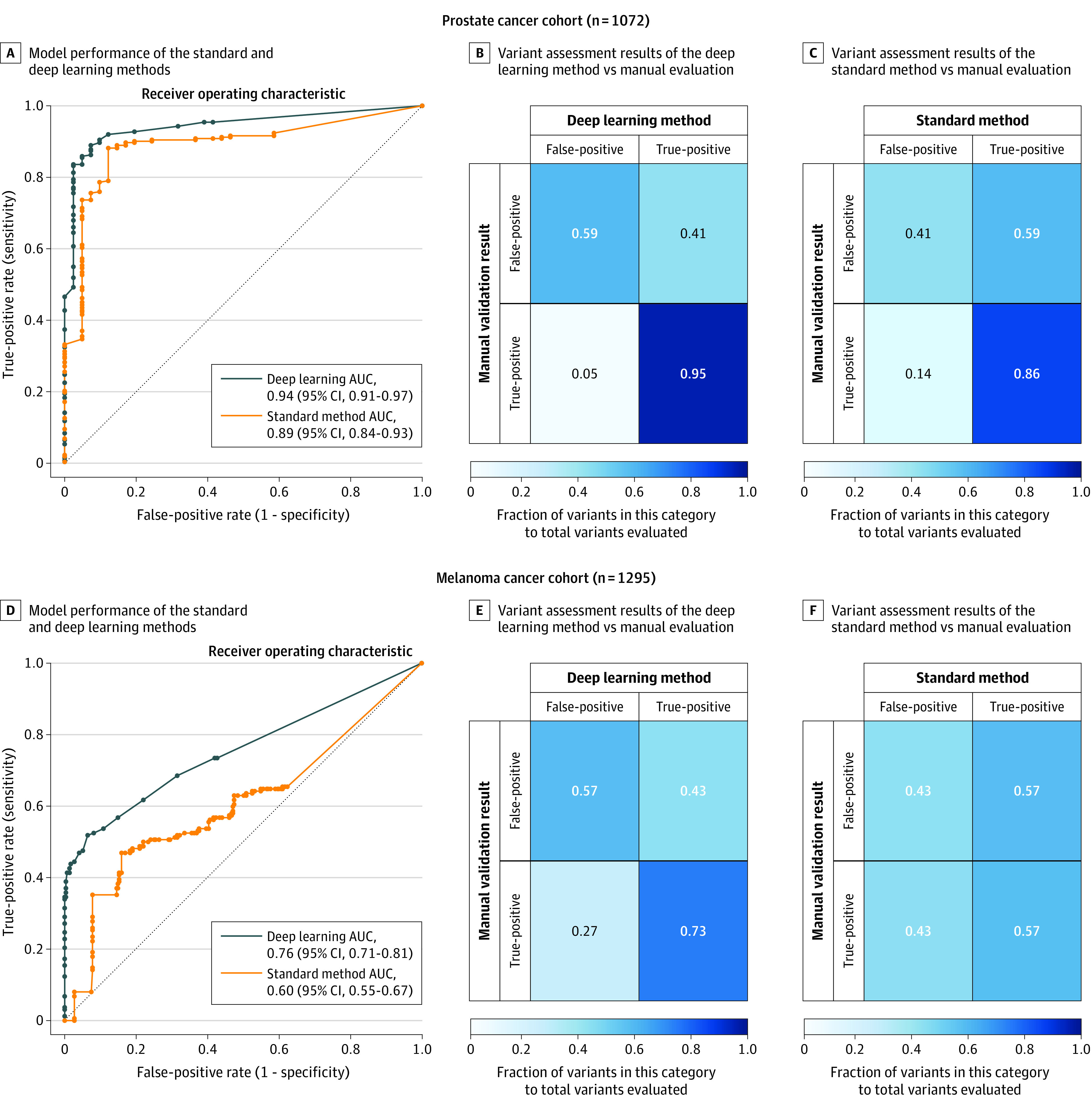
Main outcomes and measures: The primary outcomes included pathogenic variant detection performance in 118 cancer-predisposition genes estimated as sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV). The secondary outcomes were pathogenic variant detection performance in 59 genes deemed actionable by the American College of Medical Genetics and Genomics (ACMG) and 5197 clinically relevant mendelian genes. True sensitivity and true specificity could not be calculated due to lack of a criterion reference standard, but were estimated as the proportion of true-positive variants and true-negative variants, respectively, identified by each method in a reference variant set that consisted of all variants judged to be valid from either approach.
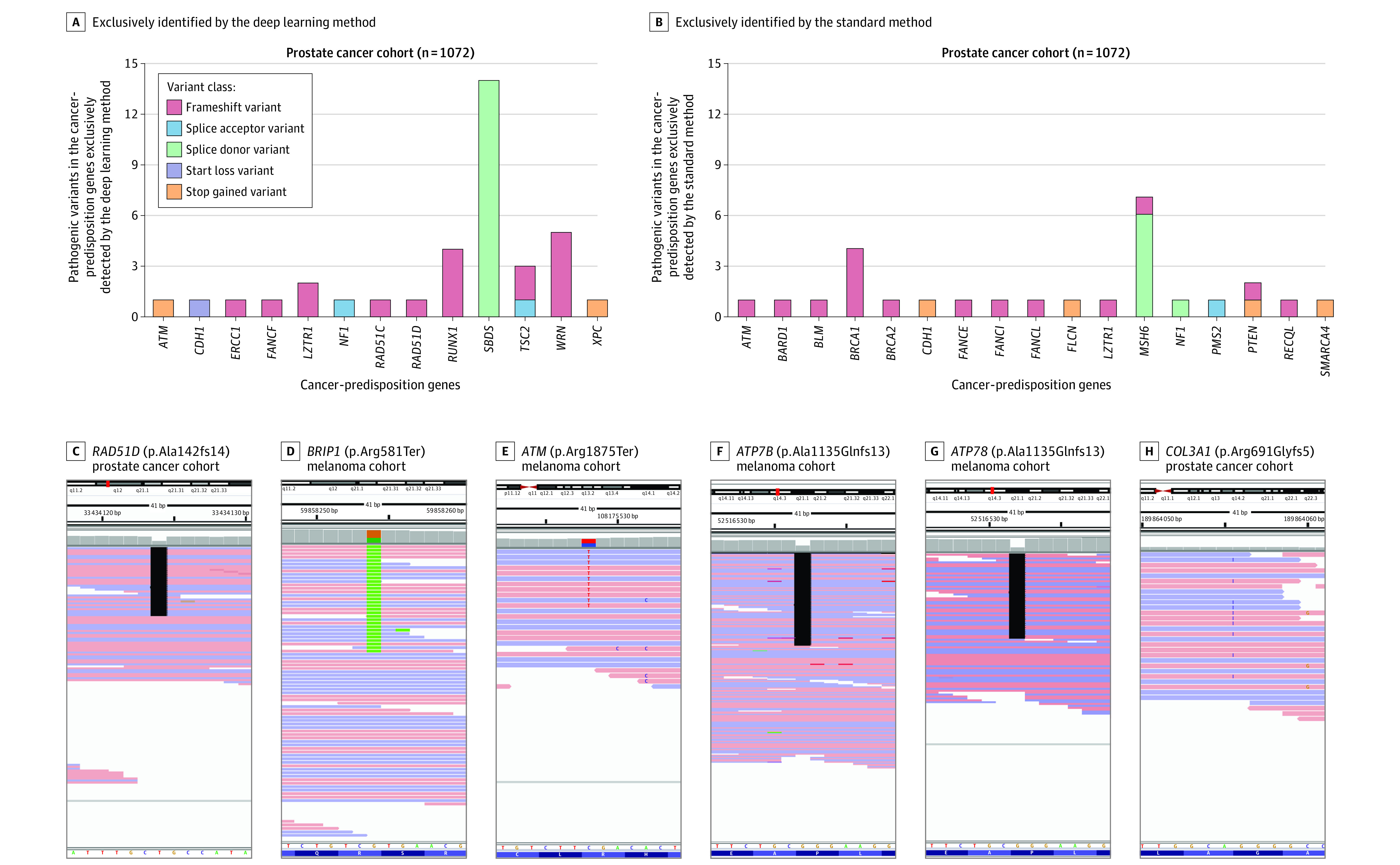
Results: The prostate cancer cohort included 1072 men (mean [SD] age at diagnosis, 63.7 [7.9] years; 857 [79.9%] with European ancestry) and the melanoma cohort included 1295 patients (mean [SD] age at diagnosis, 59.8 [15.6] years; 488 [37.7%] women; 1060 [81.9%] with European ancestry). The deep learning method identified more patients with pathogenic variants in cancer-predisposition genes than the standard method (prostate cancer: 198 vs 182; melanoma: 93 vs 74); sensitivity (prostate cancer: 94.7% vs 87.1% [difference, 7.6%; 95% CI, 2.2% to 13.1%]; melanoma: 74.4% vs 59.2% [difference, 15.2%; 95% CI, 3.7% to 26.7%]), specificity (prostate cancer: 64.0% vs 36.0% [difference, 28.0%; 95% CI, 1.4% to 54.6%]; melanoma: 63.4% vs 36.6% [difference, 26.8%; 95% CI, 17.6% to 35.9%]), PPV (prostate cancer: 95.7% vs 91.9% [difference, 3.8%; 95% CI, -1.0% to 8.4%]; melanoma: 54.4% vs 35.4% [difference, 19.0%; 95% CI, 9.1% to 28.9%]), and NPV (prostate cancer: 59.3% vs 25.0% [difference, 34.3%; 95% CI, 10.9% to 57.6%]; melanoma: 80.8% vs 60.5% [difference, 20.3%; 95% CI, 10.0% to 30.7%]). For the ACMG genes, the sensitivity of the 2 methods was not significantly different in the prostate cancer cohort (94.9% vs 90.6% [difference, 4.3%; 95% CI, -2.3% to 10.9%]), but the deep learning method had a higher sensitivity in the melanoma cohort (71.6% vs 53.7% [difference, 17.9%; 95% CI, 1.82% to 34.0%]). The deep learning method had higher sensitivity in the mendelian genes (prostate cancer: 99.7% vs 95.1% [difference, 4.6%; 95% CI, 3.0% to 6.3%]; melanoma: 91.7% vs 86.2% [difference, 5.5%; 95% CI, 2.2% to 8.8%]).
Conclusions and relevance: Among a convenience sample of 2 independent cohorts of patients with prostate cancer and melanoma, germline genetic testing using deep learning, compared with the current standard genetic testing method, was associated with higher sensitivity and specificity for detection of pathogenic variants. Further research is needed to understand the relevance of these findings with regard to clinical outcomes.
Conflict of interest statement
Figures
Comment in
-
Bioinformatics, Sequencing Accuracy, and the Credibility of Clinical Genomics.JAMA. 2020 Nov 17;324(19):1945-1947. doi: 10.1001/jama.2020.19939. JAMA. 2020. PMID: 33201189 No abstract available.
-
Urological Oncology: Prostate Cancer.J Urol. 2021 May;205(5):1515-1517. doi: 10.1097/JU.0000000000001673. Epub 2021 Feb 24. J Urol. 2021. PMID: 33625902 No abstract available.
References
-
- AlDubayan SH, Pyle LC, Gamulin M, et al. ; Regeneron Genetics Center (RGC) Research Team . Association of inherited pathogenic variants in checkpoint kinase 2 (CHEK2) with susceptibility to testicular germ cell tumors. JAMA Oncol. 2019;5(4):514-522. doi: 10.1001/jamaoncol.2018.6477 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
