

Colon cancer-specific diagnostic and prognostic biomarkers based on genome-wide abnormal DNA methylation
- PMID: 33202377
- PMCID: PMC7746390
- DOI: 10.18632/aging.103874
Colon cancer-specific diagnostic and prognostic biomarkers based on genome-wide abnormal DNA methylation
Abstract
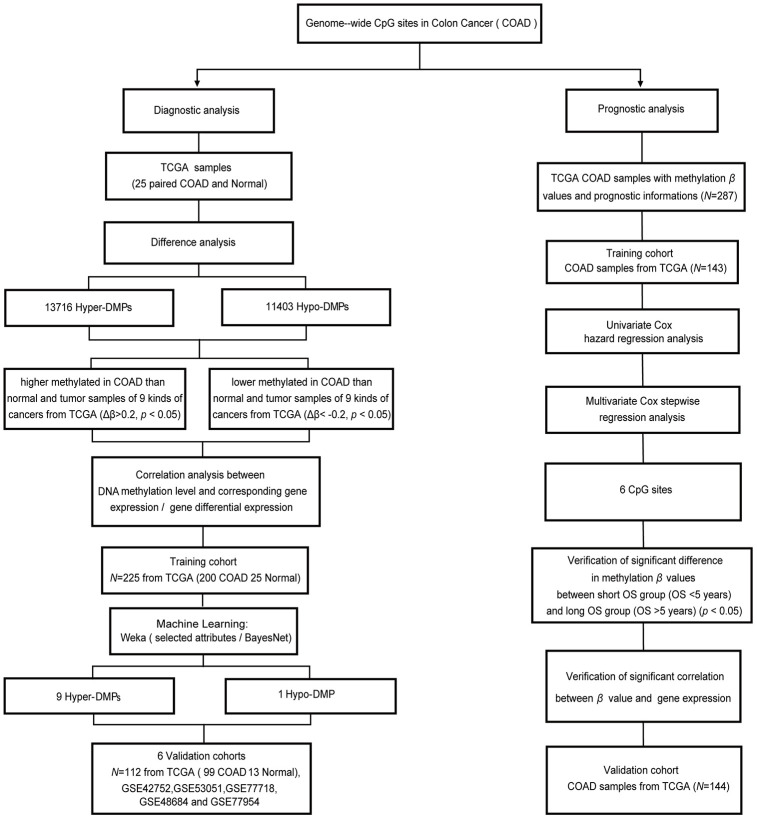
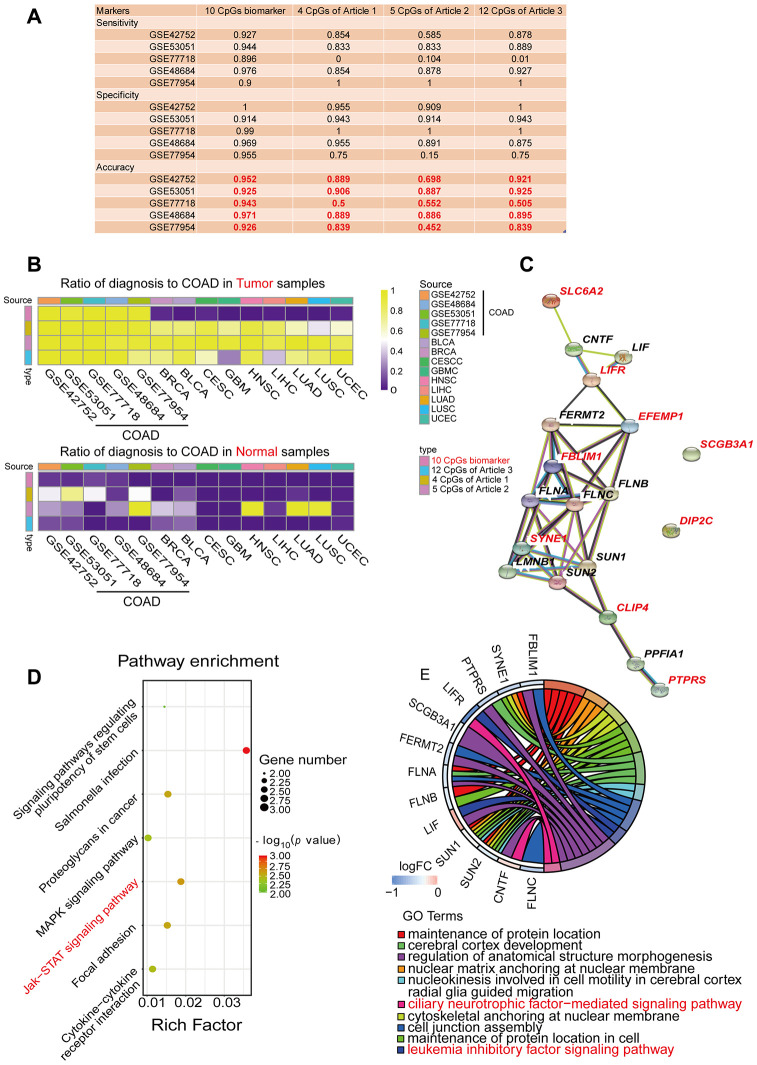
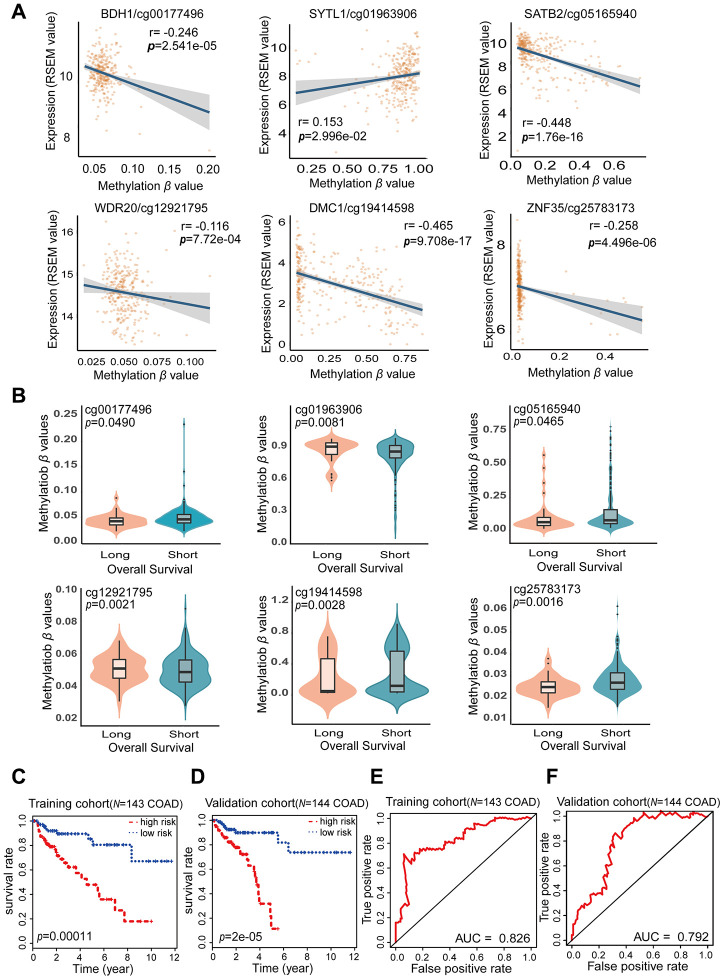
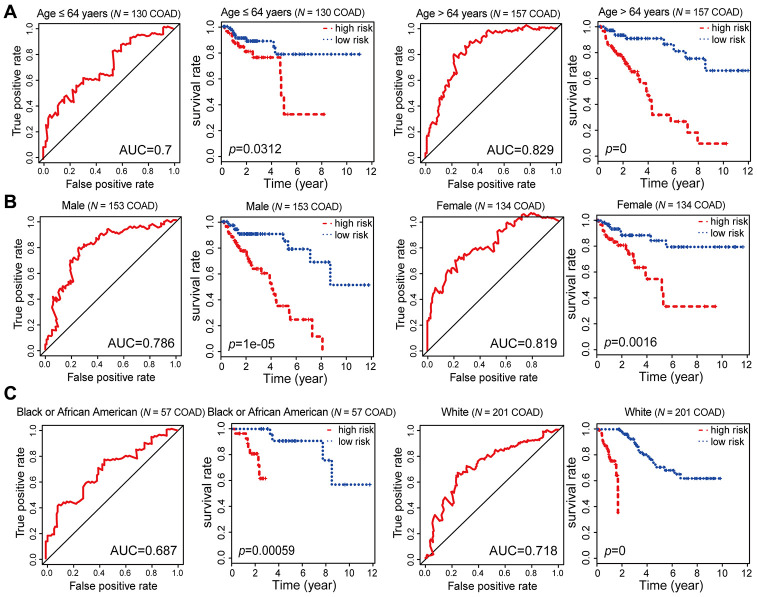
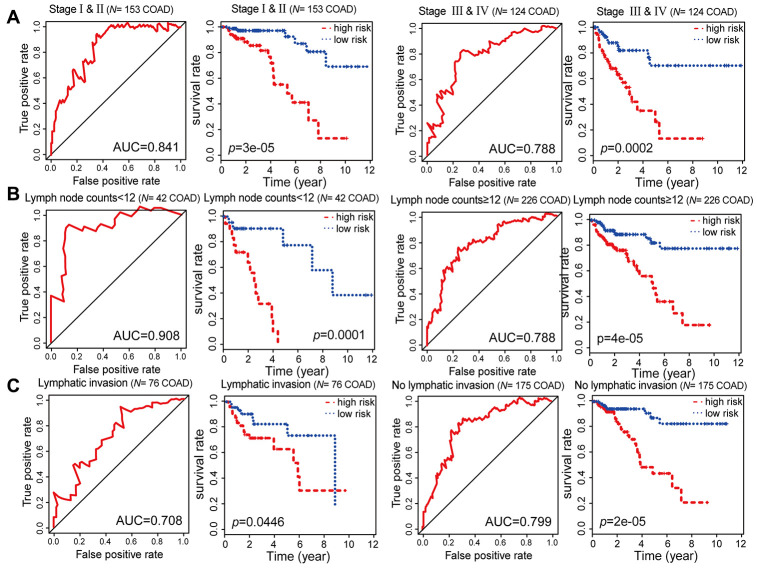
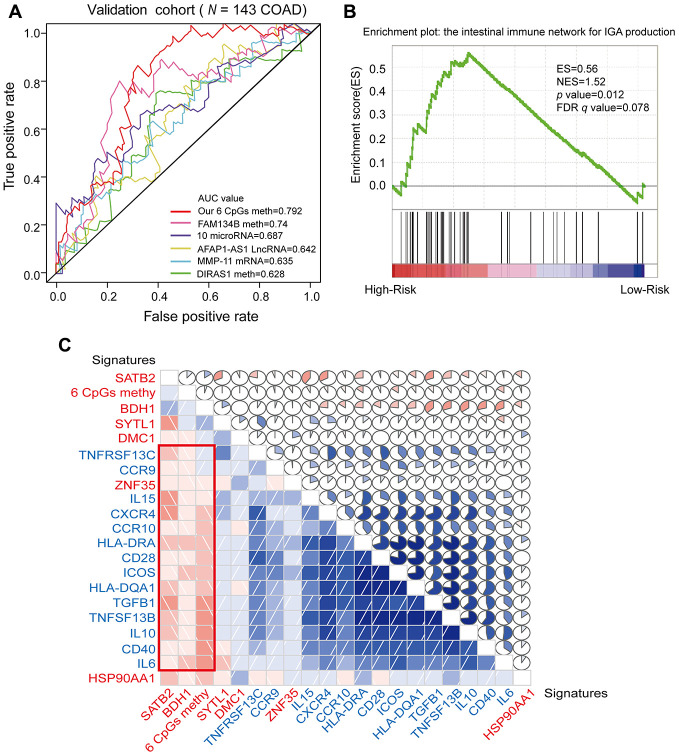
Abnormal DNA methylation is a major early contributor to colon cancer (COAD) development. We conducted a cohort-based systematic investigation of genome-wide DNA methylation using 299 COAD and 38 normal tissue samples from TCGA. Through conditional screening and machine learning with a training cohort, we identified one hypomethylated and nine hypermethylated differentially methylated CpG sites as potential diagnostic biomarkers, and used them to construct a COAD-specific diagnostic model. Unlike previous models, our model precisely distinguished COAD from nine other cancer types (e.g., breast cancer and liver cancer; error rate ≤ 0.05) and from normal tissues in the training cohort (AUC = 1). The diagnostic model was verified using a validation cohort from The Cancer Genome Atlas (AUC = 1) and five independent cohorts from the Gene Expression Omnibus (AUC ≥ 0.951). Using Cox regression analyses, we established a prognostic model based on six CpG sites in the training cohort, and verified the model in the validation cohort. The prognostic model sensitively predicted patients' survival (p ≤ 0.00011, AUC ≥ 0.792) independently of important clinicopathological characteristics of COAD (e.g., gender and age). Thus, our DNA methylation analysis provided precise biomarkers and models for the early diagnosis and prognostic evaluation of COAD.
Keywords: COAD; DMP; diagnosis; pan-cancer; prognosis.
Conflict of interest statement
Figures


Similar articles
-
Genome-wide methylation analysis identifies novel prognostic methylation markers in colon adenocarcinoma.Biomed Pharmacother. 2018 Dec;108:288-296. doi: 10.1016/j.biopha.2018.09.043. Epub 2018 Sep 15. Biomed Pharmacother. 2018. PMID: 30223100
-
Development and Validation of a Prognostic Model based on 11 E3-related Genes for Colon Cancer Patients.Curr Pharm Des. 2024;30(12):935-951. doi: 10.2174/0113816128292398240306160051. Curr Pharm Des. 2024. PMID: 38898815
-
Gene expression and methylation profiles identified CXCL3 and CXCL8 as key genes for diagnosis and prognosis of colon adenocarcinoma.J Cell Physiol. 2020 May;235(5):4902-4912. doi: 10.1002/jcp.29368. Epub 2019 Nov 10. J Cell Physiol. 2020. PMID: 31709538
-
CpG methylation signature predicts prognosis in breast cancer.Breast Cancer Res Treat. 2019 Dec;178(3):565-572. doi: 10.1007/s10549-019-05417-3. Epub 2019 Sep 13. Breast Cancer Res Treat. 2019. PMID: 31520283
-
Development and validation of a risk prediction model and nomogram for colon adenocarcinoma based on methylation-driven genes.Aging (Albany NY). 2021 Jun 28;13(12):16600-16619. doi: 10.18632/aging.203179. Epub 2021 Jun 28. Aging (Albany NY). 2021. PMID: 34182539 Free PMC article.
Cited by
-
Novel DNA methylation-based epigenetic signatures in colorectal cancer from peripheral blood leukocytes.Am J Cancer Res. 2024 May 15;14(5):2253-2271. doi: 10.62347/MXWJ1398. eCollection 2024. Am J Cancer Res. 2024. PMID: 38859857 Free PMC article.
-
Expression mode and prognostic value of FXYD family members in colon cancer.Aging (Albany NY). 2021 Jul 15;13(14):18404-18422. doi: 10.18632/aging.203290. Epub 2021 Jul 15. Aging (Albany NY). 2021. PMID: 34270462 Free PMC article.
-
Genome-wide DNA methylation profiles of colorectal tumors in Lynch syndrome and familial adenomatous polyposis.Clin Epigenetics. 2025 Aug 2;17(1):137. doi: 10.1186/s13148-025-01940-x. Clin Epigenetics. 2025. PMID: 40753397 Free PMC article.
-
Conserved methylation signatures associate with the tumor immune microenvironment and immunotherapy response.Genome Med. 2024 Apr 2;16(1):47. doi: 10.1186/s13073-024-01318-3. Genome Med. 2024. PMID: 38566132 Free PMC article.
-
Prognostic Score Model Based on Ten Differentially Methylated Genes for Predicting Clinical Outcomes in Patients with Adenocarcinoma of the Colon.Cancer Manag Res. 2021 Jun 28;13:5113-5125. doi: 10.2147/CMAR.S312085. eCollection 2021. Cancer Manag Res. 2021. PMID: 34234555 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
