

Somatic Epigenetic Silencing of RIPK3 Inactivates Necroptosis and Contributes to Chemoresistance in Malignant Mesothelioma
- PMID: 33203643
- PMCID: PMC7887036
- DOI: 10.1158/1078-0432.CCR-18-3683
Somatic Epigenetic Silencing of RIPK3 Inactivates Necroptosis and Contributes to Chemoresistance in Malignant Mesothelioma
Abstract
Purpose: Receptor-interacting protein kinase 3 (RIPK3) phosphorylates effector molecule MLKL to trigger necroptosis. Although RIPK3 loss is seen in several human cancers, its role in malignant mesothelioma is unknown. This study aimed to determine whether RIPK3 functions as a potential tumor suppressor to limit development of malignant mesothelioma.
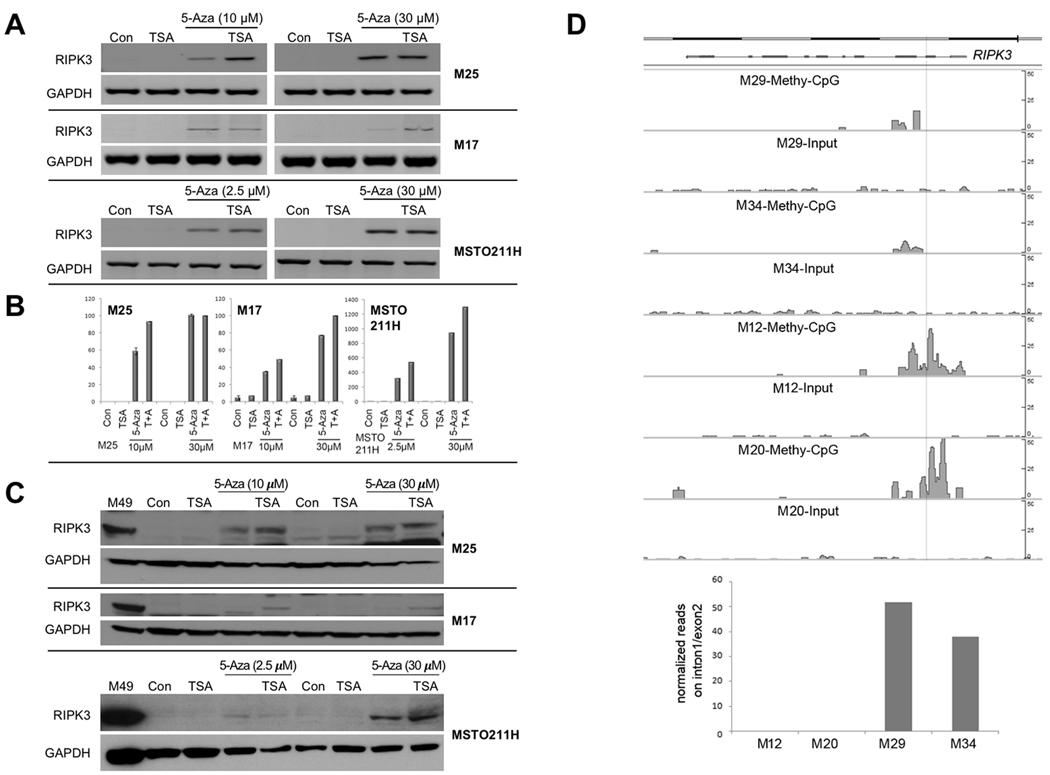
Experimental design: RIPK3 expression was examined in 66 malignant mesothelioma tumors and cell lines. Promoter methylation and DNMT1 siRNA studies were performed to assess the mode of RIPK3 silencing in RIPK3-deficient malignant mesothelioma cells. Restoration of RIPK3 expression in RIPK3-negative malignant mesothelioma cells, either by treatment with 5-aza-2'-deoxycytidine or lentiviral expression of cDNA, was performed to assess effects on cell viability, necrosis, and chemosensitization.
Results: Loss of RIPK3 expression was observed in 42/66 (63%) primary malignant mesotheliomas and malignant mesothelioma cell lines, and RT-PCR analysis demonstrated that downregulation occurs at the transcriptional level, consistent with epigenetic silencing. RIPK3-negative malignant mesothelioma cells treated with 5-aza-2'-deoxycytidine resulted in reexpression of RIPK3 and chemosensitization. Ectopic expression of RIPK3 also resulted in chemosensitization and led to necroptosis, the latter demonstrated by phosphorylation of downstream target MLKL and confirmed by rescue experiments. Mining of RIPK3 expression and survival outcomes among patients with malignant mesothelioma available from The Cancer Genome Atlas repository revealed that promoter methylation of RIPK3 is associated with reduced RIPK3 expression and poor prognosis.
Conclusions: These data suggest that RIPK3 acts as a tumor suppressor in malignant mesothelioma by triggering necroptosis and that epigenetic silencing of RIPK3 by DNA methylation impairs necroptosis and contributes to chemoresistance and poor survival in this incurable disease.
©2020 American Association for Cancer Research.
Conflict of interest statement
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest with regard to this work.
Figures






References
-
- Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee W-C, Altomare DA, et al. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res 1994;54:5547–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
