

Chicoric Acid Ameliorates Nonalcoholic Fatty Liver Disease via the AMPK/Nrf2/NF κ B Signaling Pathway and Restores Gut Microbiota in High-Fat-Diet-Fed Mice
- PMID: 33204402
- PMCID: PMC7657699
- DOI: 10.1155/2020/9734560
Chicoric Acid Ameliorates Nonalcoholic Fatty Liver Disease via the AMPK/Nrf2/NF κ B Signaling Pathway and Restores Gut Microbiota in High-Fat-Diet-Fed Mice
Abstract
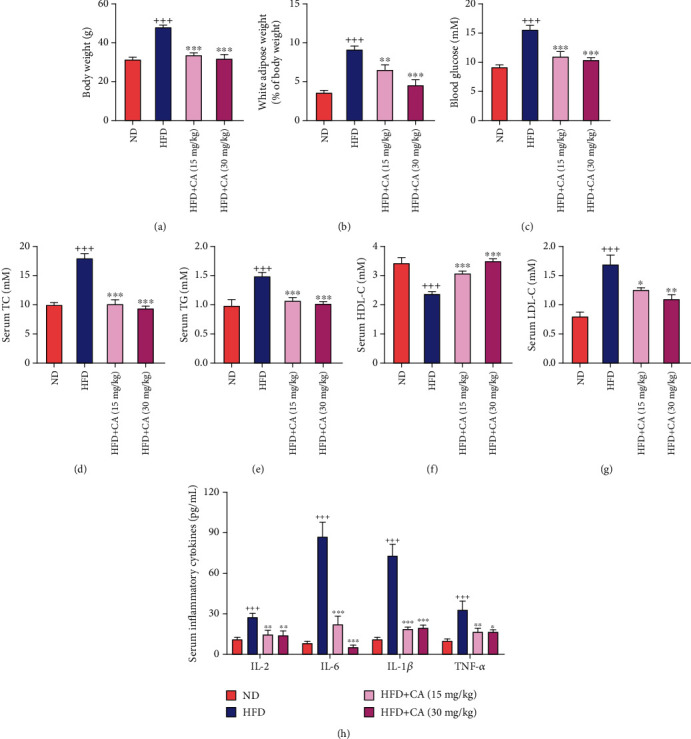
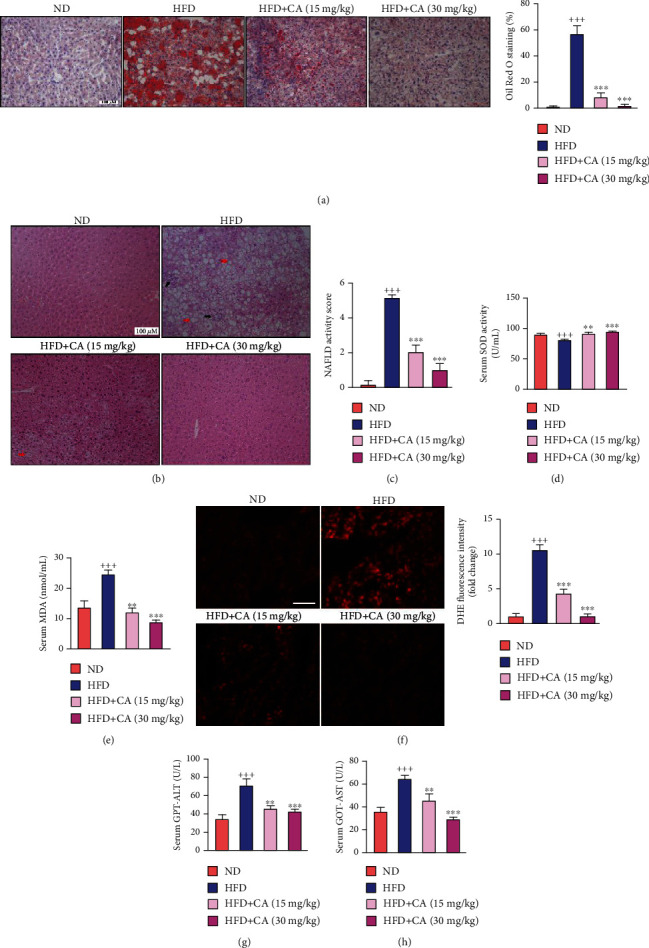
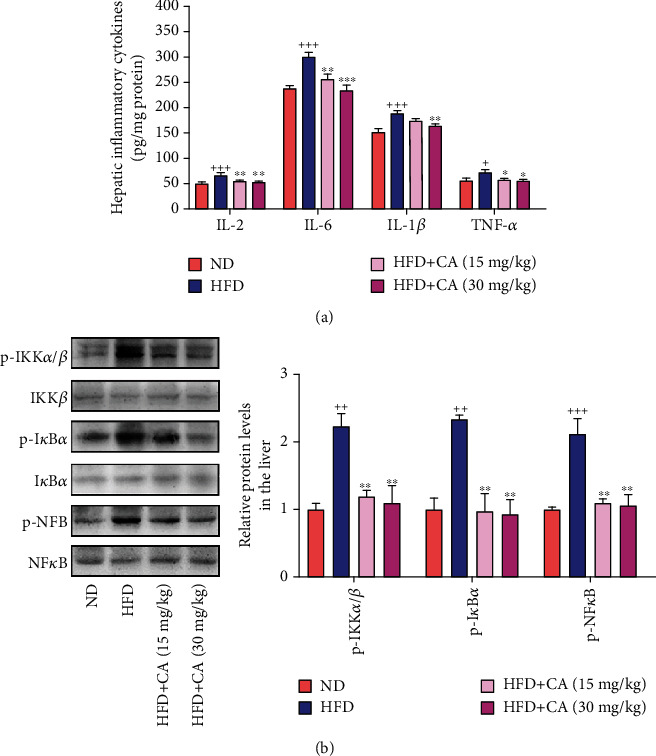
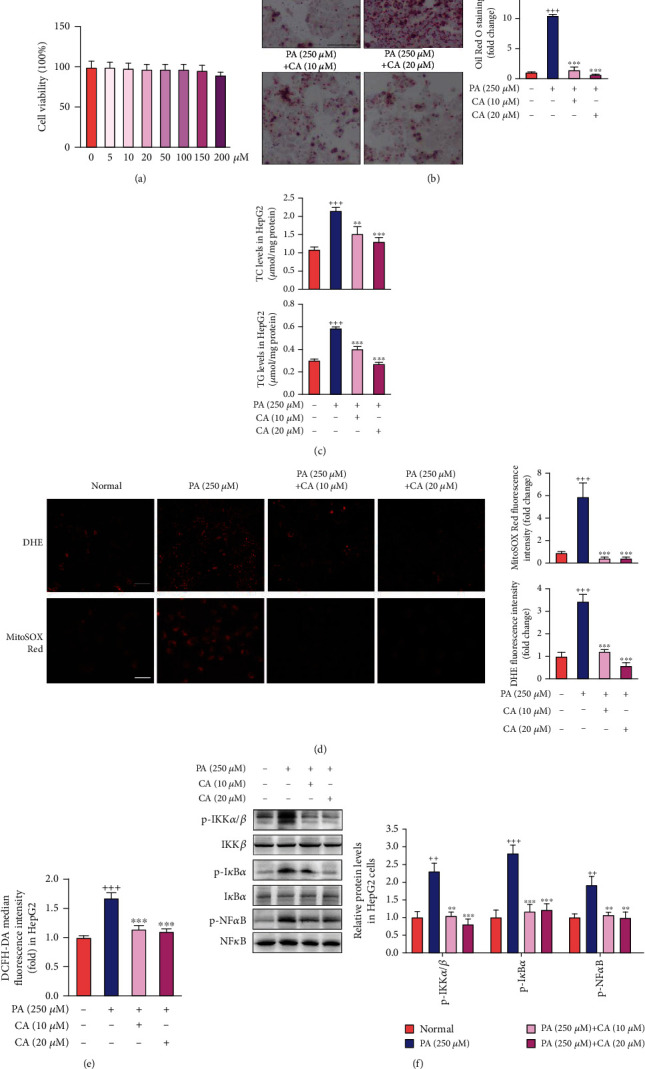
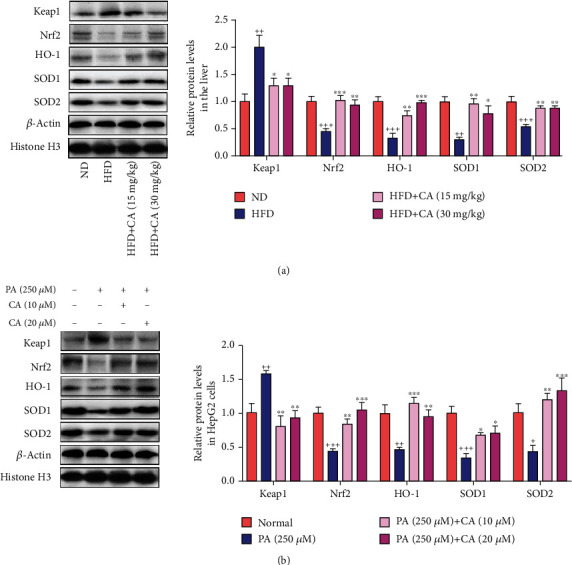
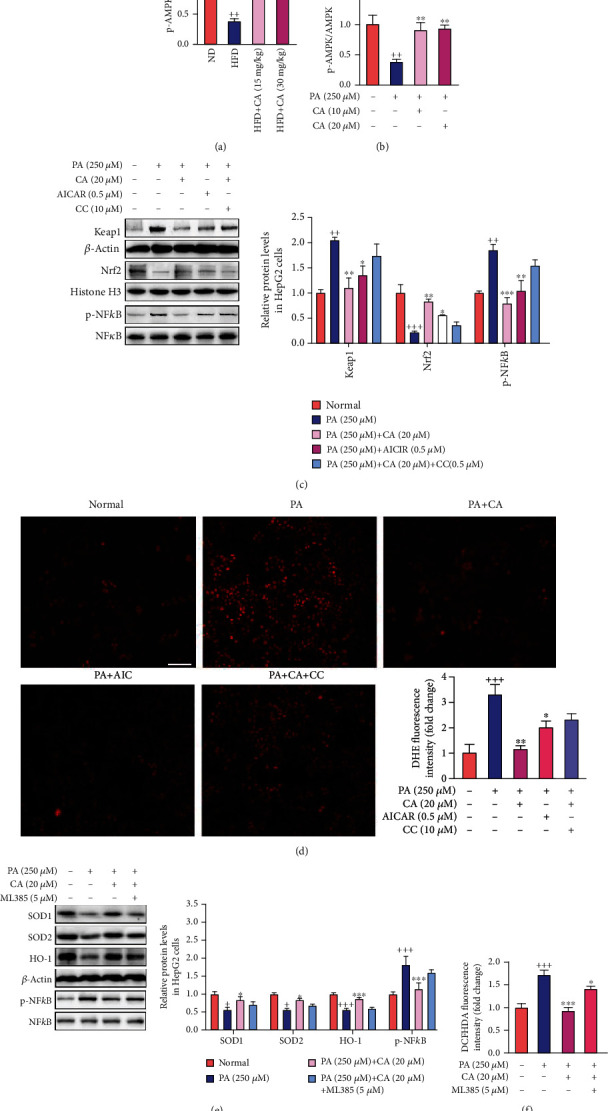
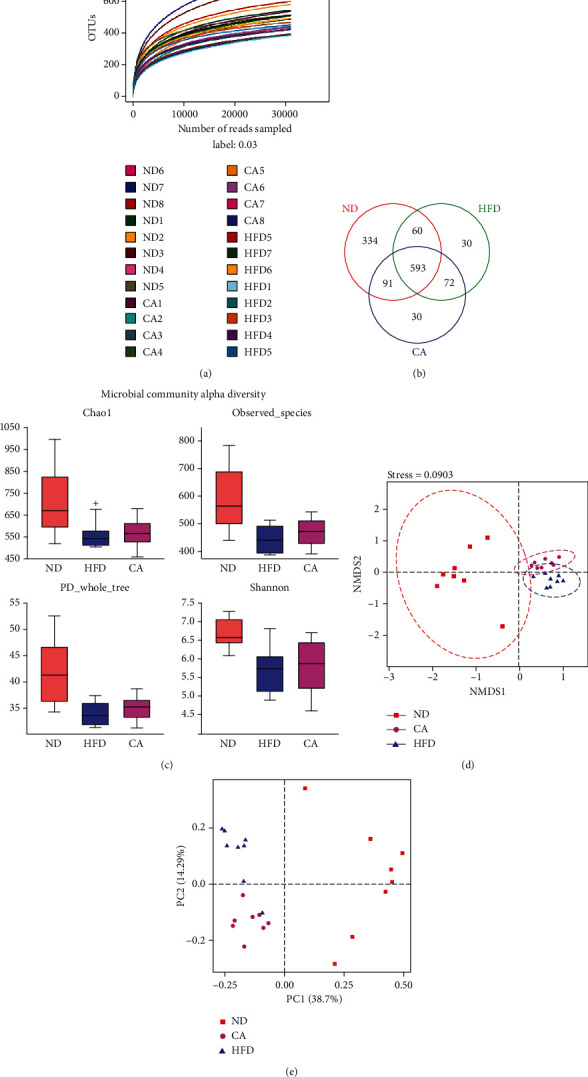
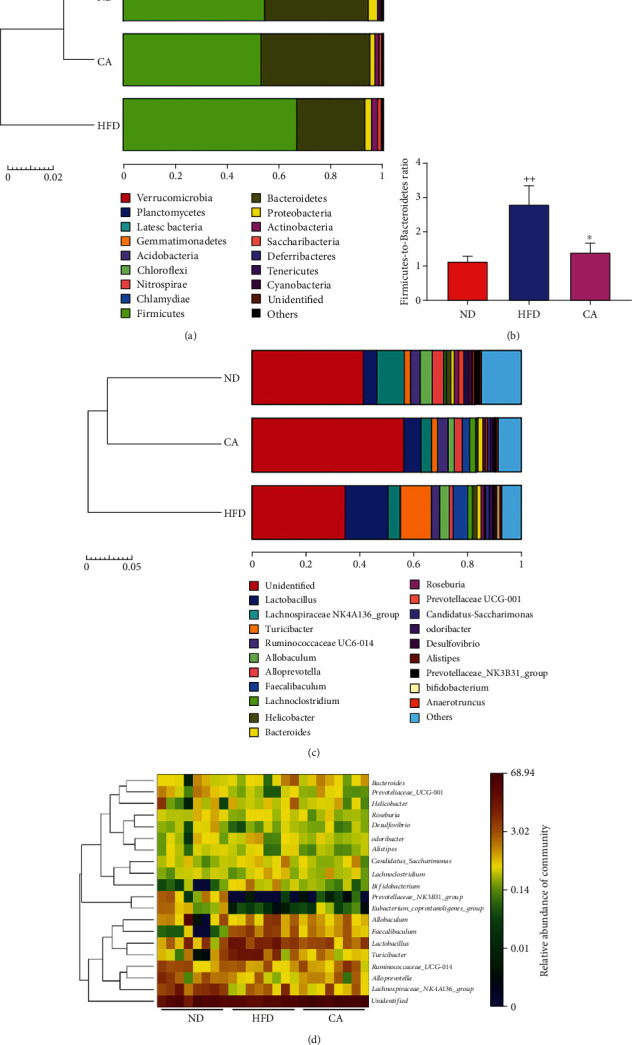
This study examines the effects of chicoric acid (CA) on nonalcoholic fatty liver disease (NAFLD) in high-fat-diet- (HFD-) fed C57BL/6 mice. CA treatment decreased body weight and white adipose weight, mitigated hyperglycemia and dyslipidemia, and reduced hepatic steatosis in HFD-fed mice. Moreover, CA treatment reversed HFD-induced oxidative stress and inflammation both systemically and locally in the liver, evidenced by the decreased serum malondialdehyde (MDA) abundance, increased serum superoxide dismutase (SOD) activity, lowered in situ reactive oxygen species (ROS) in the liver, decreased serum and hepatic inflammatory cytokine levels, and reduced hepatic inflammatory cell infiltration in HFD-fed mice. In addition, CA significantly reduced lipid accumulation and oxidative stress in palmitic acid- (PA-) treated HepG2 cells. In particular, we identified AMPK as an activator of Nrf2 and an inactivator of NFκB. CA upregulated AMPK phosphorylation, the nuclear protein level of Nrf2, and downregulated NFκB protein level both in HFD mice and PA-treated HepG2 cells. Notably, AMPK inhibitor compound C blocked the regulation of Nrf2 and NFκB, as well as ROS overproduction mediated by CA in PA-treated HepG2 cells, while AMPK activator AICAR mimicked the effects of CA. Similarly, Nrf2 inhibitor ML385 partly blocked the regulation of antioxidative genes and ROS overproduction by CA in PA-treated HepG2 cells. Interestingly, high-throughput pyrosequencing of 16S rRNA suggested that CA could increase Firmicutes-to-Bacteroidetes ratio and modify gut microbial composition towards a healthier microbial profile. In summary, CA plays a preventative role in the amelioration of oxidative stress and inflammation via the AMPK/Nrf2/NFκB signaling pathway and shapes gut microbiota in HFD-induced NAFLD.
Copyright © 2020 Xiaoqin Ding et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Wanless I. R., Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Seminars in Liver Disease. 2004;24(1):99–106. doi: 10.1055/s-2004-823104. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
