

A Random Matrix Theory Approach to Denoise Single-Cell Data
- PMID: 33205104
- PMCID: PMC7660363
- DOI: 10.1016/j.patter.2020.100035
A Random Matrix Theory Approach to Denoise Single-Cell Data
Abstract
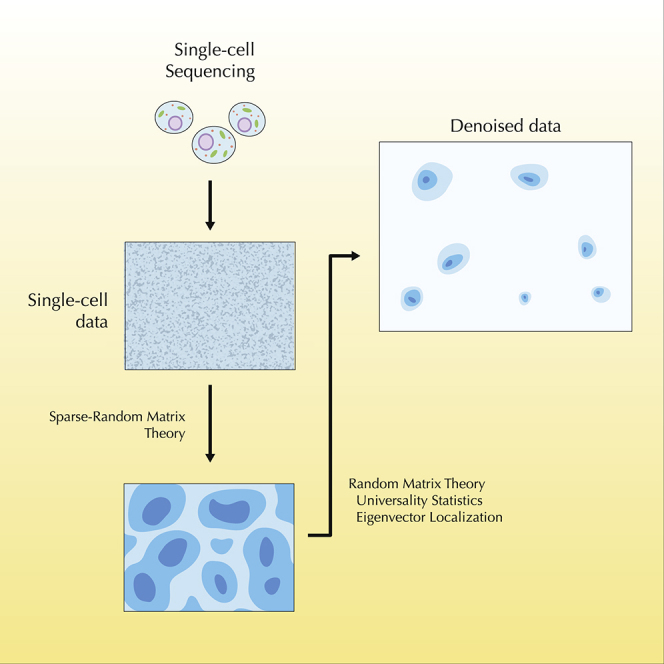
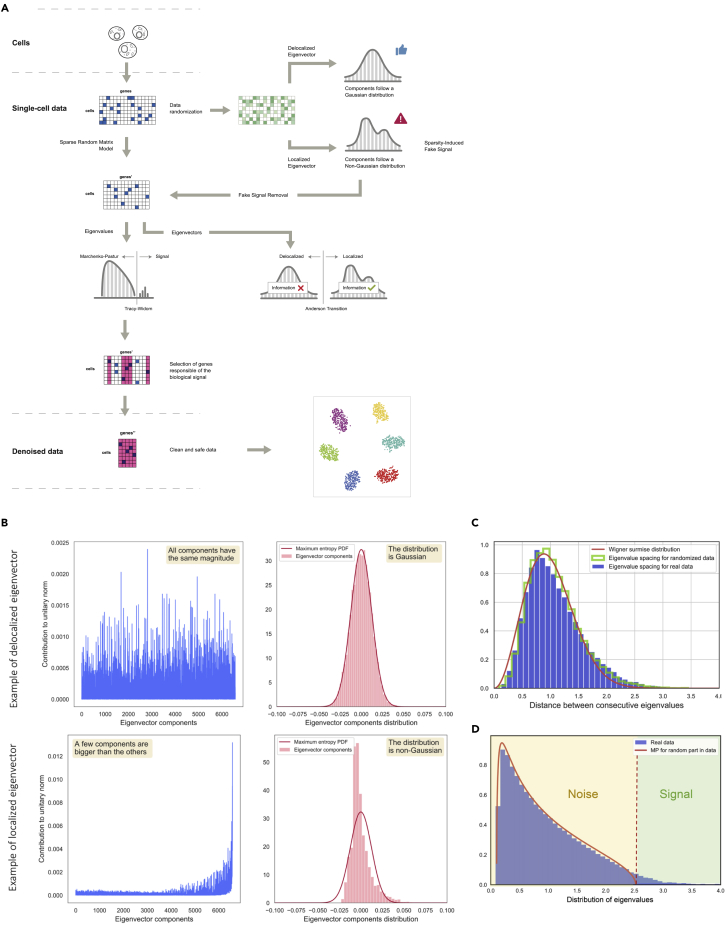
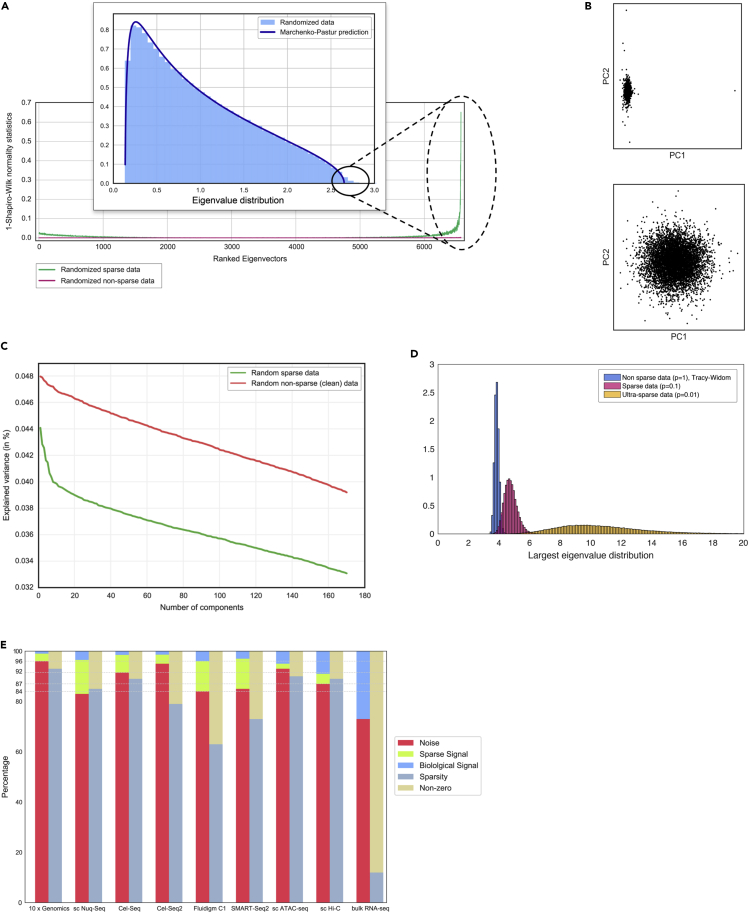
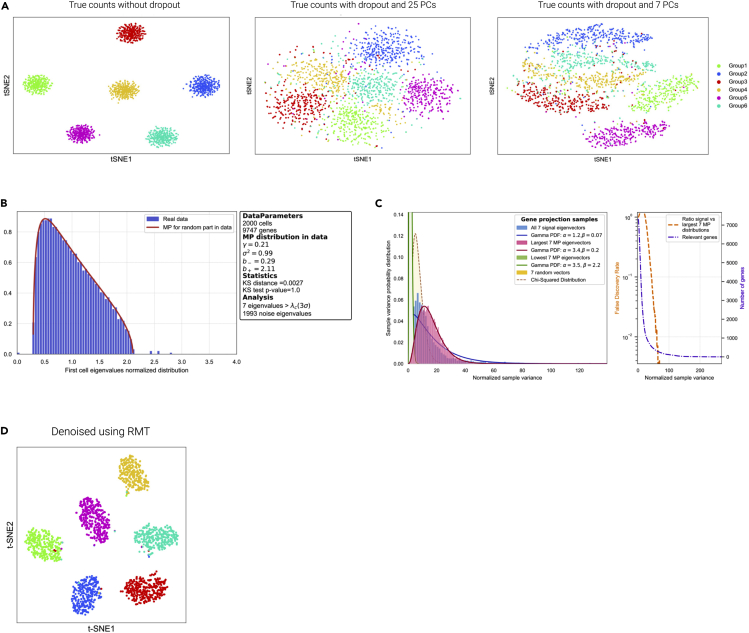
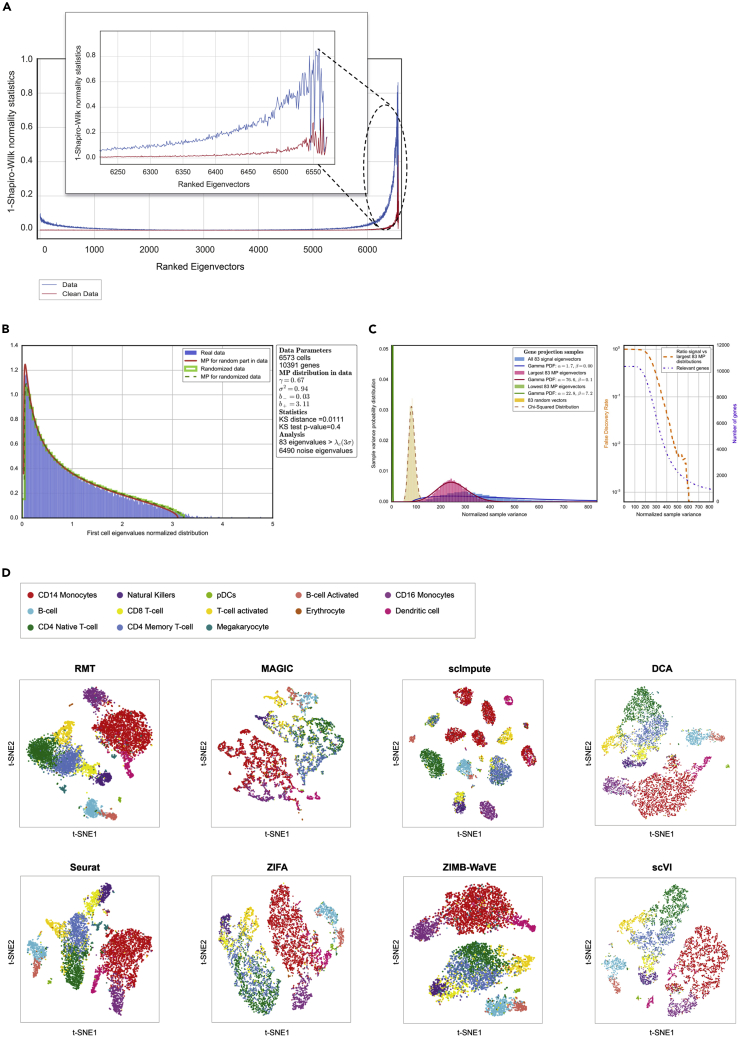
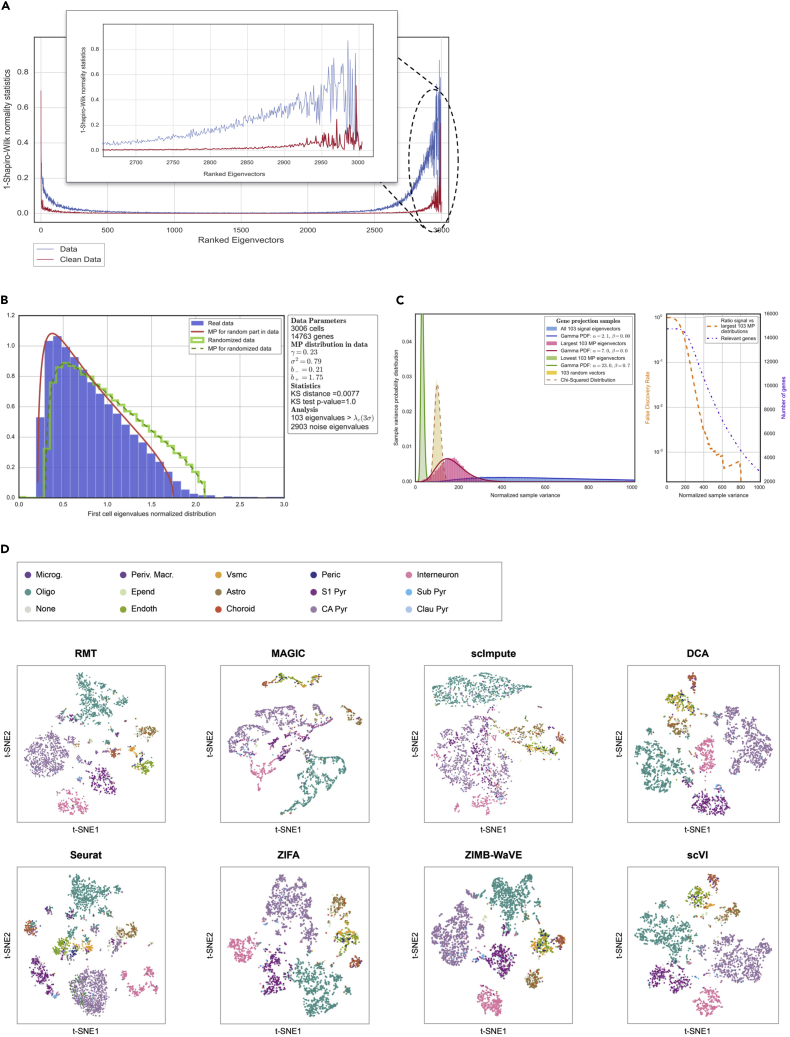
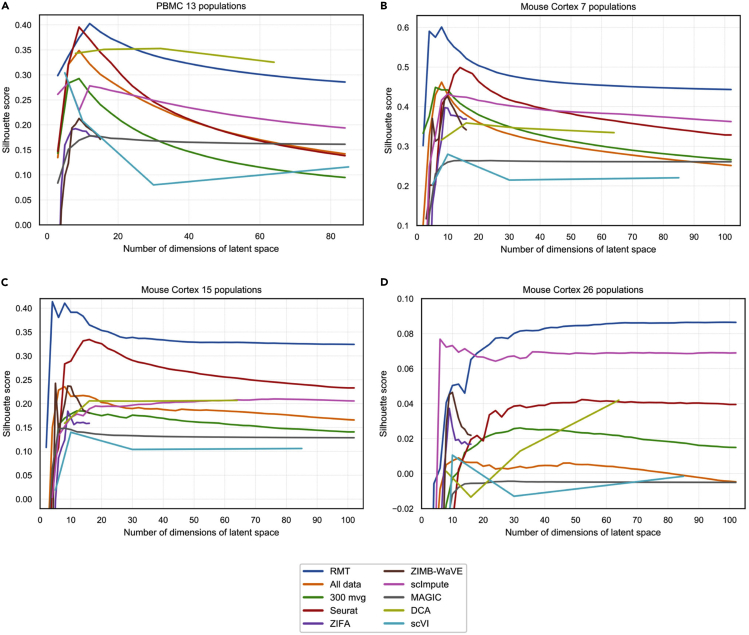
Single-cell technologies provide the opportunity to identify new cellular states. However, a major obstacle to the identification of biological signals is noise in single-cell data. In addition, single-cell data are very sparse. We propose a new method based on random matrix theory to analyze and denoise single-cell sequencing data. The method uses the universal distributions predicted by random matrix theory for the eigenvalues and eigenvectors of random covariance/Wishart matrices to distinguish noise from signal. In addition, we explain how sparsity can cause spurious eigenvector localization, falsely identifying meaningful directions in the data. We show that roughly 95% of the information in single-cell data is compatible with the predictions of random matrix theory, about 3% is spurious signal induced by sparsity, and only the last 2% reflects true biological signal. We demonstrate the effectiveness of our approach by comparing with alternative techniques in a variety of examples with marked cell populations.
Keywords: denoising; eigenvector localization; random matrix theory; single cell; sparsity; universality.
© 2020 The Author(s).
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
