

Genomic and transcriptomic correlates of Richter transformation in chronic lymphocytic leukemia
- PMID: 33206936
- PMCID: PMC8163497
- DOI: 10.1182/blood.2020005650
Genomic and transcriptomic correlates of Richter transformation in chronic lymphocytic leukemia
Abstract
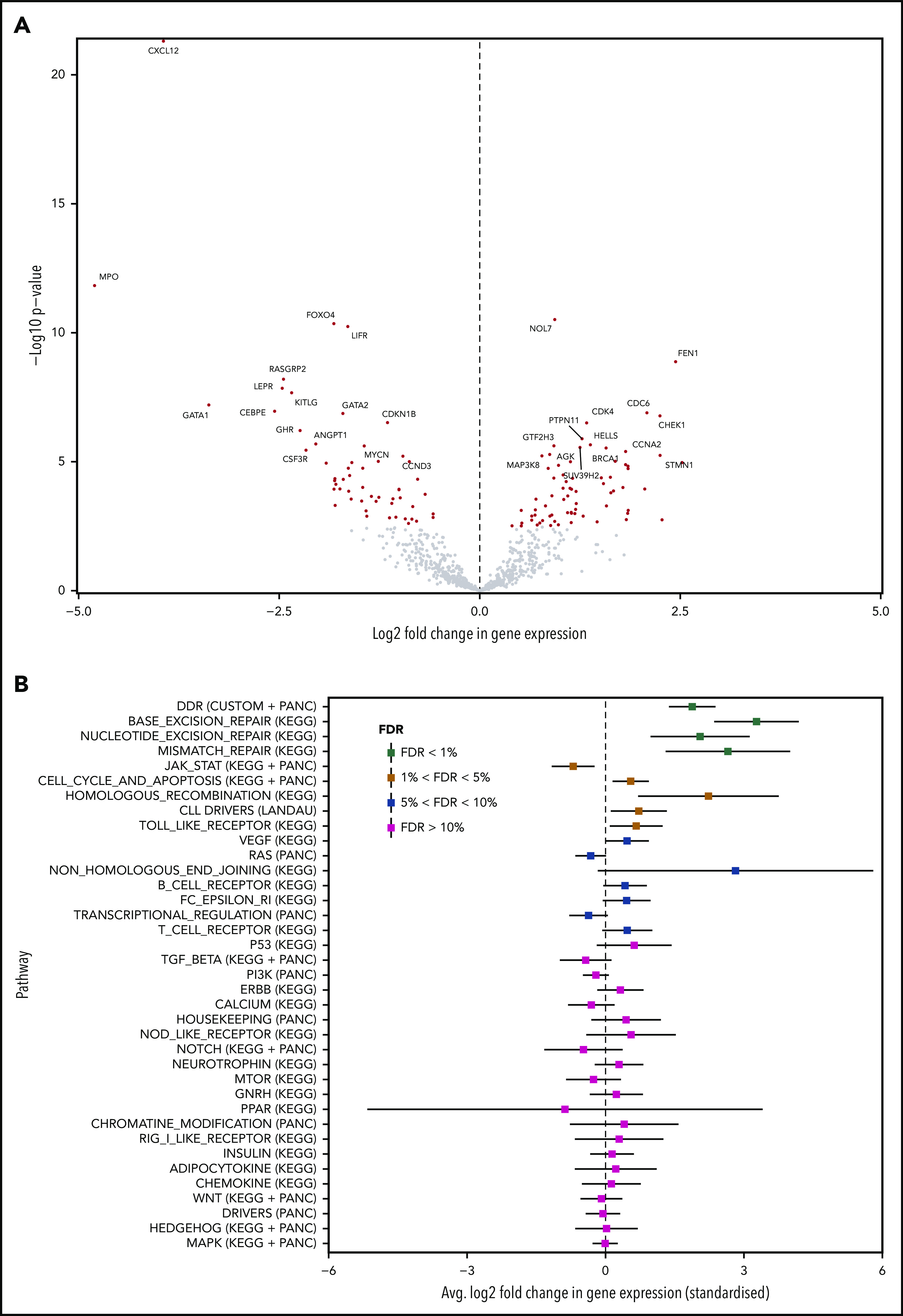
The transformation of chronic lymphocytic leukemia (CLL) to high-grade B-cell lymphoma is known as Richter syndrome (RS), a rare event with dismal prognosis. In this study, we conducted whole-genome sequencing (WGS) of paired circulating CLL (PB-CLL) and RS biopsies (tissue-RS) from 17 patients recruited into a clinical trial (CHOP-O). We found that tissue-RS was enriched for mutations in poor-risk CLL drivers and genes in the DNA damage response (DDR) pathway. In addition, we identified genomic aberrations not previously implicated in RS, including the protein tyrosine phosphatase receptor (PTPRD) and tumor necrosis factor receptor-associated factor 3 (TRAF3). In the noncoding genome, we discovered activation-induced cytidine deaminase-related and unrelated kataegis in tissue-RS affecting regulatory regions of key immune-regulatory genes. These include BTG2, CXCR4, NFATC1, PAX5, NOTCH-1, SLC44A5, FCRL3, SELL, TNIP2, and TRIM13. Furthermore, differences between the global mutation signatures of pairs of PB-CLL and tissue-RS samples implicate DDR as the dominant mechanism driving transformation. Pathway-based clonal deconvolution analysis showed that genes in the MAPK and DDR pathways demonstrate high clonal-expansion probability. Direct comparison of nodal-CLL and tissue-RS pairs from an independent cohort confirmed differential expression of the same pathways by RNA expression profiling. Our integrated analysis of WGS and RNA expression data significantly extends previous targeted approaches, which were limited by the lack of germline samples, and it facilitates the identification of novel genomic correlates implicated in RS transformation, which could be targeted therapeutically. Our results inform the future selection of investigative agents for a UK clinical platform study. This trial was registered at www.clinicaltrials.gov as #NCT03899337.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: A.S. received honoraria from Gilead, Janssen, Roche, and AbbVie, and received nonrestricted educational grants from Gilead and Janssen. T.A.E. received honoraria from Roche, Janssen, AbbVie, Gilead, and AstraZeneca, and received travel fees to scientific conferences from Takeda. N.A. received speaker fees from Gilead. The remaining authors declare no competing financial interests.
Figures







Comment in
-
Differential transcriptomic profiling in ibrutinib-naïve versus ibrutinib-resistant Richter syndrome.Hematol Oncol. 2022 Apr;40(2):302-306. doi: 10.1002/hon.2950. Epub 2021 Dec 3. Hematol Oncol. 2022. PMID: 34806797 No abstract available.
References
-
- Tsimberidou AM, Keating MJ. Richter’s transformation in chronic lymphocytic leukemia. Semin Oncol. 2006;33(2):250-256. - PubMed
-
- Rossi D, Spina V, Deambrogi C, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391-3401. - PubMed
-
- Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761-2772. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
