

Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights
- PMID: 33208169
- PMCID: PMC7677848
- DOI: 10.1186/s12931-020-01570-2
Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights
Abstract
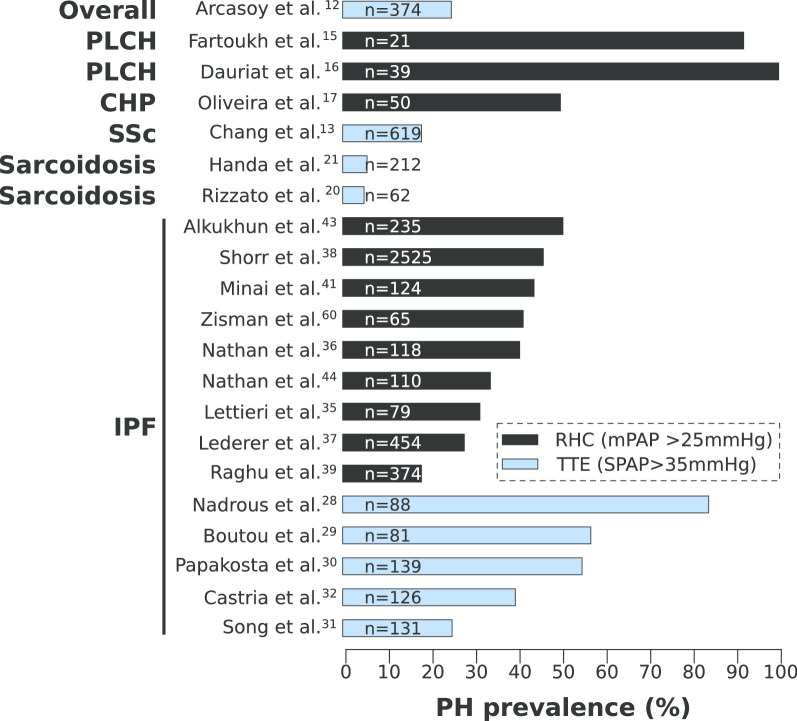
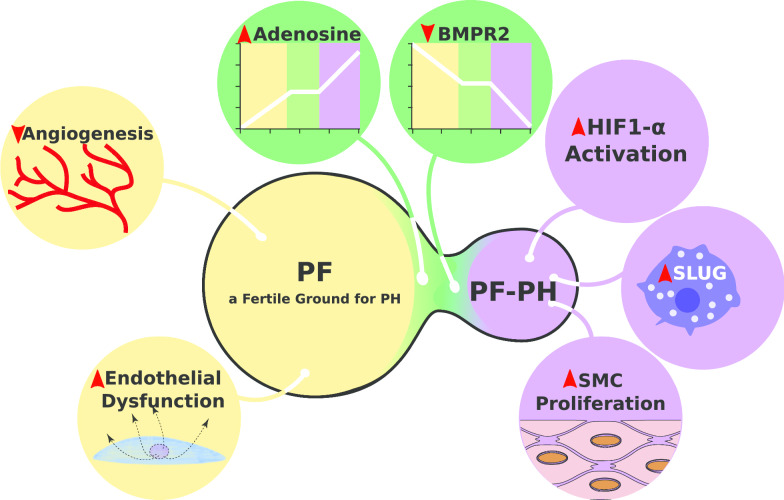
Pulmonary hypertension (PH) developing secondarily in pulmonary fibrosis (PF) patients (PF-PH) is a frequent co-morbidity. The high prevalence of PH in PF patients is very concerning since the presence of PH is a strong predictor of mortality in PF patients. Until recently, PH was thought to arise solely from fibrotic destruction of the lung parenchyma, leading to hypoxic vasoconstriction and loss of vascular bed density. Thus, potential cellular and molecular dysregulation of vascular remodeling as a driver of PF-PH has been under-investigated. The recent demonstrations that there is no correlation between the severity of the fibrosis and development of PH, along with the finding that significant vascular histological and molecular differences exist between patients with and without PH have shifted the etiological paradigm of PF-PH. This review aims to provide a comprehensive translational overview of PH in PF patients from clinical diagnosis and outcome to the latest understanding of the histology and molecular pathophysiology of PF-PH.
Keywords: Pulmonary fibrosis; Pulmonary hypertension; Vascular diseases.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
