

Functional Characterization of Circulating Mumps Viruses with Stop Codon Mutations in the Small Hydrophobic Protein
- PMID: 33208518
- PMCID: PMC7677008
- DOI: 10.1128/mSphere.00840-20
Functional Characterization of Circulating Mumps Viruses with Stop Codon Mutations in the Small Hydrophobic Protein
Abstract
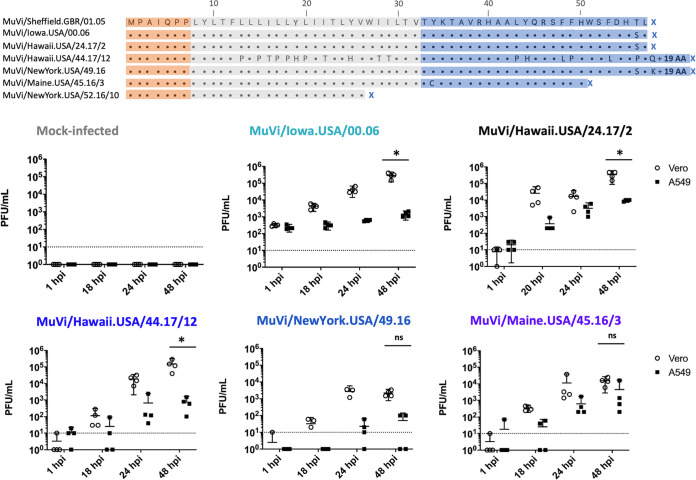
Between 2015 and 2017, routine molecular surveillance in the United States detected multiple mumps viruses (MuVs) with mutations in the small hydrophobic (SH) gene compared to a reference virus of the same genotype. These mutations include an unusual pattern of uracil-to-cytosine hypermutations and other mutations resulting in the generation of premature stop codons or disruption of the canonical stop codon. The mumps virus SH protein may serve as a virulence factor, based on evidence that it inhibits apoptosis and innate immune signaling in vitro and that recombinant viruses that do not express the SH protein are attenuated in an animal model. In this study, mumps viruses bearing variant SH sequences were isolated from contemporary outbreak samples to evaluate the impact of the observed mutations on SH protein function. All isolates with variant SH sequences replicated in interferon-competent cells with no evidence of attenuation. Furthermore, all SH-variant viruses retained the ability to abrogate induction of NF-κB-mediated innate immune signaling in infected cells. Ectopic expression of variant mumps SH genes is consistent with findings from infection experiments, indicating that the observed abrogation of signaling was not mediated by other viral factors that may modulate innate immune signaling. Molecular surveillance is an important public health tool for monitoring the diversity of circulating mumps viruses and can provide insights into determinants of disease. These findings, in turn, will inform studies employing reverse genetics to elucidate the specific mechanisms of MuV pathogenesis and potential impacts of observed sequence variants on infectivity, fitness, and virulence.IMPORTANCE Mumps virus (MuV) outbreaks occur in the United States despite high coverage with measles, mumps, rubella (MMR) vaccine. Routine genotyping of laboratory-confirmed mumps cases has been practiced in the United States since 2006 to enhance mumps surveillance. This study reports the detection of unusual mutations in the small hydrophobic (SH) protein of contemporary laboratory-confirmed mumps cases and is the first to describe the impact of such mutations on SH protein function. These mutations are predicted to profoundly alter the amino acid sequence of the SH protein, which has been shown to antagonize host innate immune responses; however, they were neither associated with defects in virus replication nor attenuated protein function in vitro, consistent with detection in clinical specimens. A better understanding of the forces governing mumps virus sequence diversity and of the functional consequences of mutations in viral proteins is important for maintaining robust capacity for mumps detection and disease control.
Keywords: SH protein; altered termination; genomics; host-pathogen interactions; molecular epidemiology; mumps virus; next-generation sequencing; paramyxovirus; surveillance studies; vaccine; vaccine preventable.
Copyright © 2020 Stinnett et al.
Figures
Similar articles
-
Discrimination of mumps virus small hydrophobic gene deletion effects from gene translation effects on virus virulence.J Virol. 2011 Jun;85(12):6082-5. doi: 10.1128/JVI.02686-10. Epub 2011 Apr 6. J Virol. 2011. PMID: 21471236 Free PMC article.
-
Role of Small Hydrophobic Protein of J Paramyxovirus in Virulence.J Virol. 2018 Sep 26;92(20):e00653-18. doi: 10.1128/JVI.00653-18. Print 2018 Oct 15. J Virol. 2018. PMID: 30068647 Free PMC article.
-
Genetic characterization of mumps viruses associated with the resurgence of mumps in the United States: 2015-2017.Virus Res. 2020 May;281:197935. doi: 10.1016/j.virusres.2020.197935. Epub 2020 Mar 16. Virus Res. 2020. PMID: 32194138
-
Unique Tropism and Entry Mechanism of Mumps Virus.Viruses. 2021 Sep 1;13(9):1746. doi: 10.3390/v13091746. Viruses. 2021. PMID: 34578327 Free PMC article. Review.
-
Measles-mumps-rubella vaccine and autistic spectrum disorder: report from the New Challenges in Childhood Immunizations Conference convened in Oak Brook, Illinois, June 12-13, 2000.Pediatrics. 2001 May;107(5):E84. doi: 10.1542/peds.107.5.e84. Pediatrics. 2001. PMID: 11331734 Review.
Cited by
-
ADAR Editing in Viruses: An Evolutionary Force to Reckon with.Genome Biol Evol. 2021 Nov 5;13(11):evab240. doi: 10.1093/gbe/evab240. Genome Biol Evol. 2021. PMID: 34694399 Free PMC article. Review.
-
Small hydrophobic (SH) proteins of Pneumoviridae and Paramyxoviridae: small but mighty.J Virol. 2024 Sep 17;98(9):e0080924. doi: 10.1128/jvi.00809-24. Epub 2024 Aug 23. J Virol. 2024. PMID: 39177356 Free PMC article. Review.
References
-
- McNall RJ, Wharton AK, Anderson R, Clemmons N, Lopareva EN, Gonzalez C, Espinosa A, Probert WS, Hacker JK, Liu G, Garfin J, Strain AK, Boxrud D, Bryant PW, St George K, Davis T, Griesser RH, Shult P, Bankamp B, Hickman CJ, Wroblewski K, Rota PA. 2020. Genetic characterization of mumps viruses associated with the resurgence of mumps in the United States: 2015–2017. Virus Res 281:197935. doi:10.1016/j.virusres.2020.197935. - DOI - PubMed
-
- World Health Organization. 2012. Mumps virus nomenclature update. Wkly Epidemiol Rec 87:217–224. - PubMed
-
- Dayan GH, Quinlisk MP, Parker AA, Barskey AE, Harris ML, Schwartz JM, Hunt K, Finley CG, Leschinsky DP, O'Keefe AL, Clayton J, Kightlinger LK, Dietle EG, Berg J, Kenyon CL, Goldstein ST, Stokley SK, Redd SB, Rota PA, Rota J, Bi D, Roush SW, Bridges CB, Santibanez TA, Parashar U, Bellini WJ, Seward JF. 2008. Recent resurgence of mumps in the United States. N Engl J Med 358:1580–1589. doi:10.1056/NEJMoa0706589. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
