

Evolutionary study of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as an emerging coronavirus: Phylogenetic analysis and literature review
- PMID: 33210477
- PMCID: PMC7753621
- DOI: 10.1002/vms3.394
Evolutionary study of COVID-19, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as an emerging coronavirus: Phylogenetic analysis and literature review
Abstract
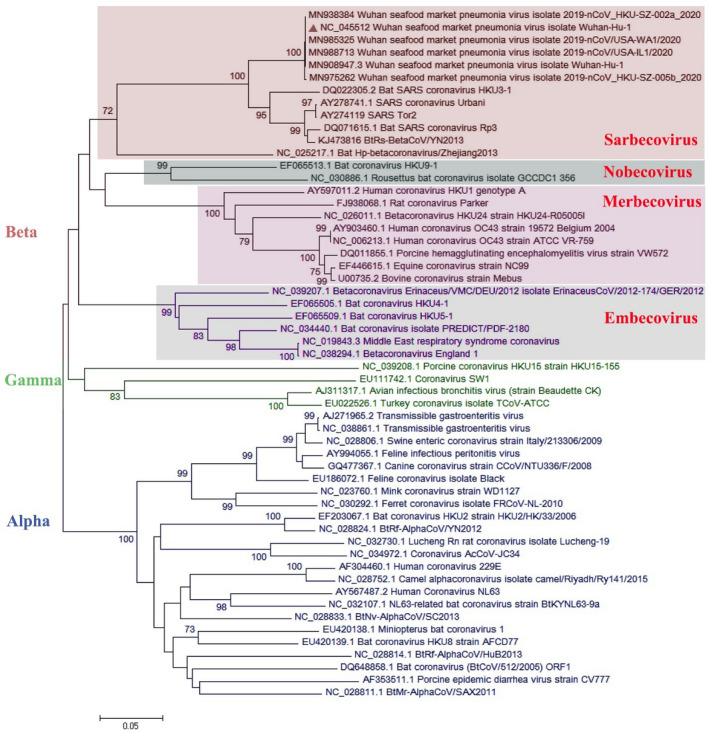
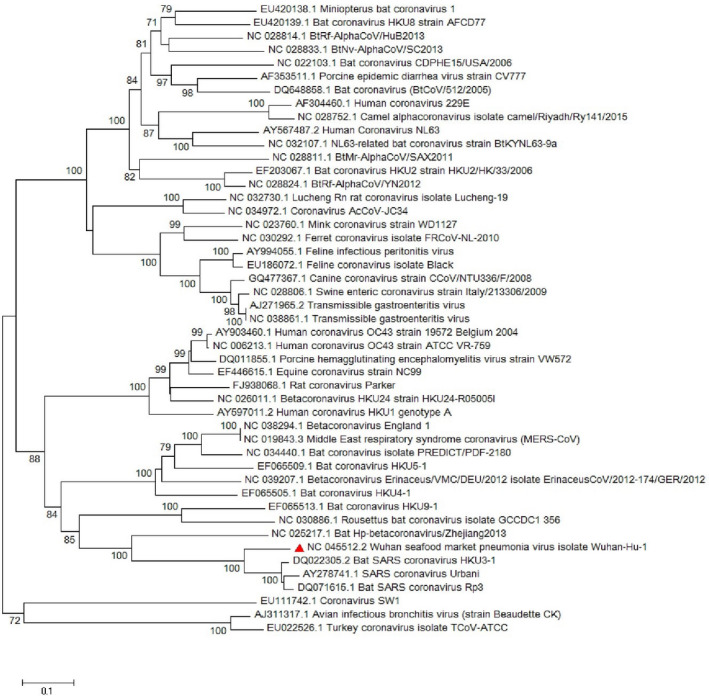
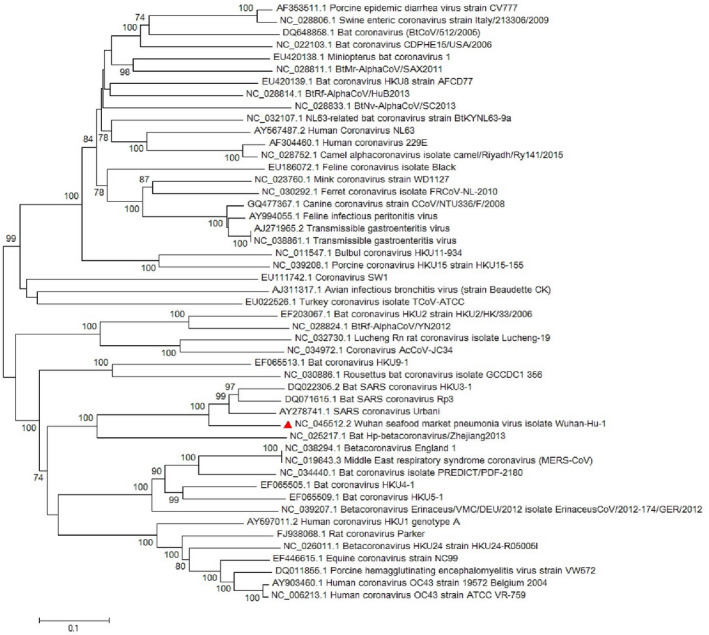
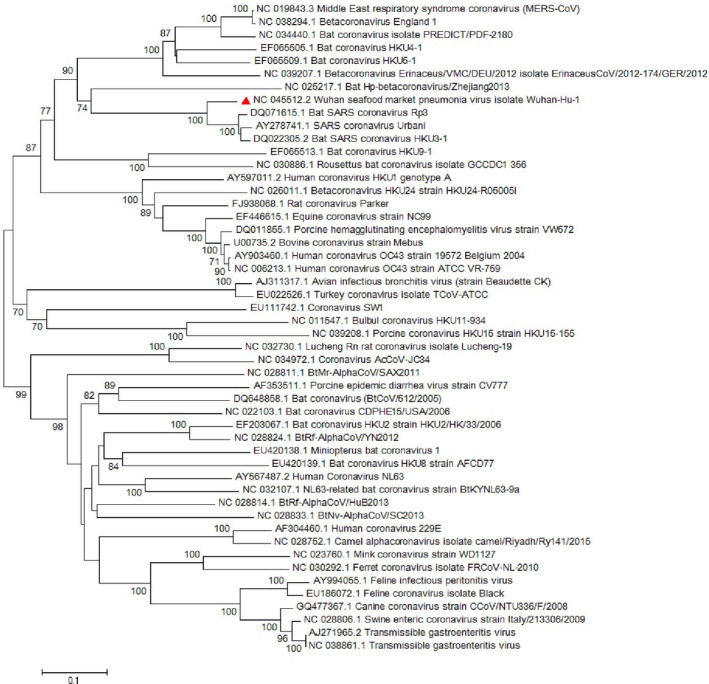
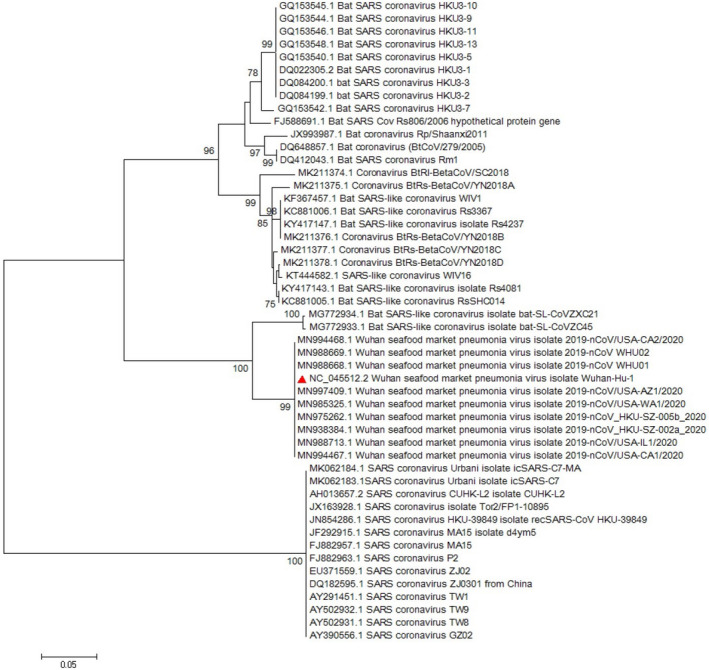
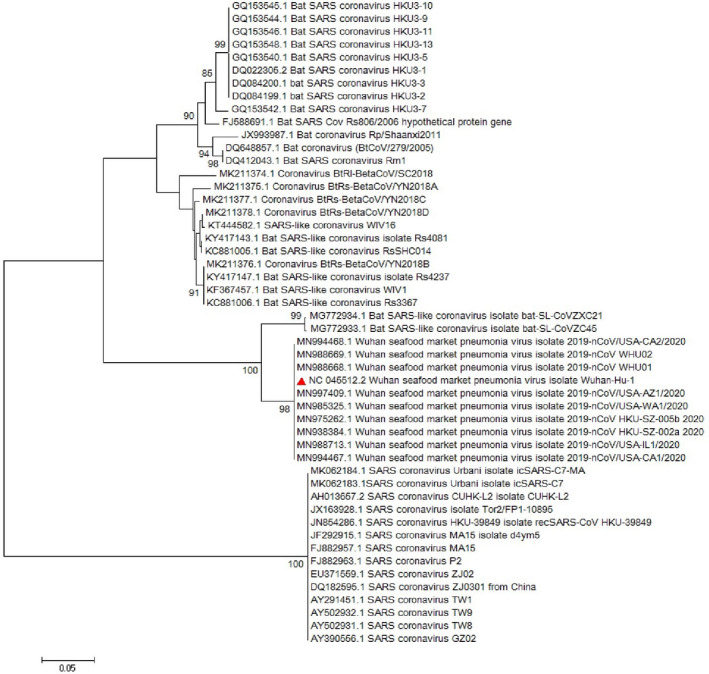
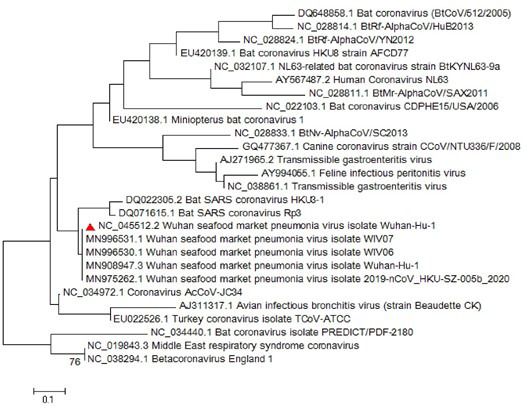
Since emerging coronaviruses have always become a human health concern globally especially severe acute respiratory syndrome coronavirus 2 (SARS-CoV) and Middle East respiratory syndrome coronavirus and a novel coronavirus was introduced in Wuhan, China, in December 2019 (called SARS-CoV-2), many researchers focused on its epidemics, virological and clinical features. SARS-CoV-2 is classified as Betacoronaviruses genus and Sarbecovirus subgenus (lineage B). The virus shows a great similarity with SARS-CoV and bat SARS-like coronaviruses. In this study, we evaluate SARS-CoV-2 virus phylogeny and evolution by using current virus and related sequences.
Keywords: COVID-19; evolutionary study; phylogenetic analysis; severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
© 2020 The Authors Veterinary Medicine and Science Published by John Wiley & Sons Ltd.
Conflict of interest statement
None declared.
Figures


References
-
- Bai, Y. , Jiang, D. , Lon, J. R. , Chen, X. , Hu, M. , Lin, S. , Chen, Z. , Wang, X. , Meng, Y. , & Dua, H. (2020). Comprehensive evolution and molecular characteristics of a large number of SARS‐CoV‐2 genomes reveal its epidemic trends. International Journal of Infectious Diseases, 100, 164–173. 10.1016/j.ijid.2020.08.066 - DOI - PMC - PubMed
-
- Borchardt, R. A. , & Rolston, K. V. (2012). Respiratory tract infections: Emerging viral pathogens. Journal of the American Academy of Physician Assistants, 25(10), 19–20. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
