

Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy
- PMID: 33212010
- PMCID: PMC8077737
- DOI: 10.1016/j.cell.2020.10.036
Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy
Abstract
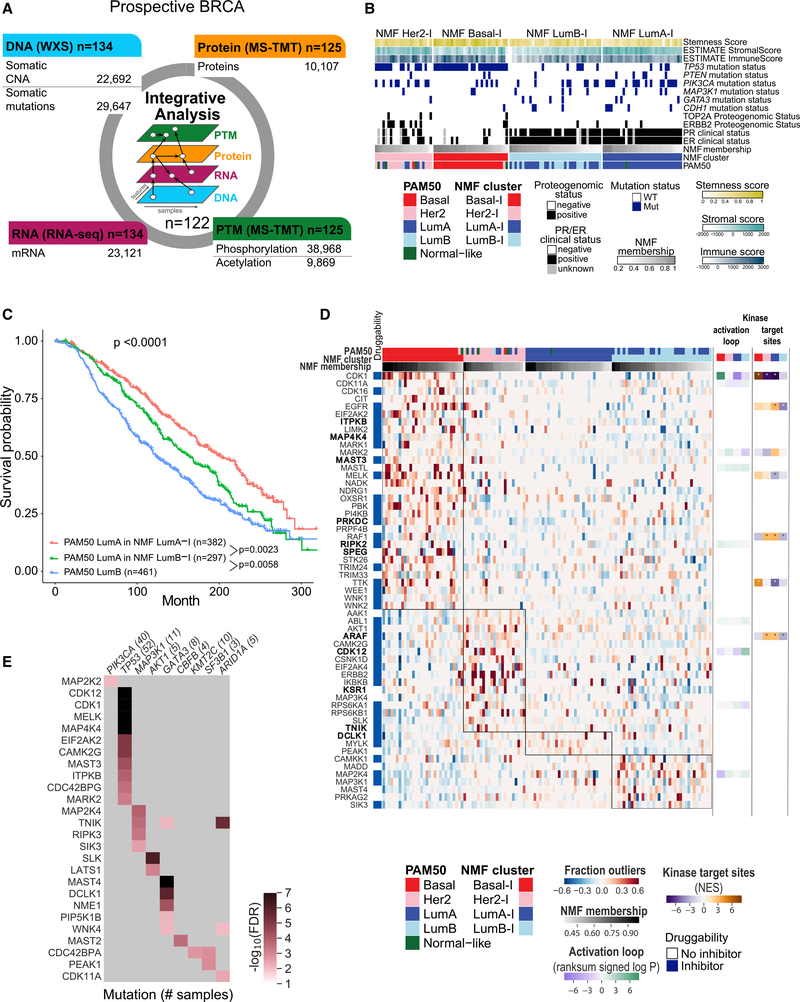
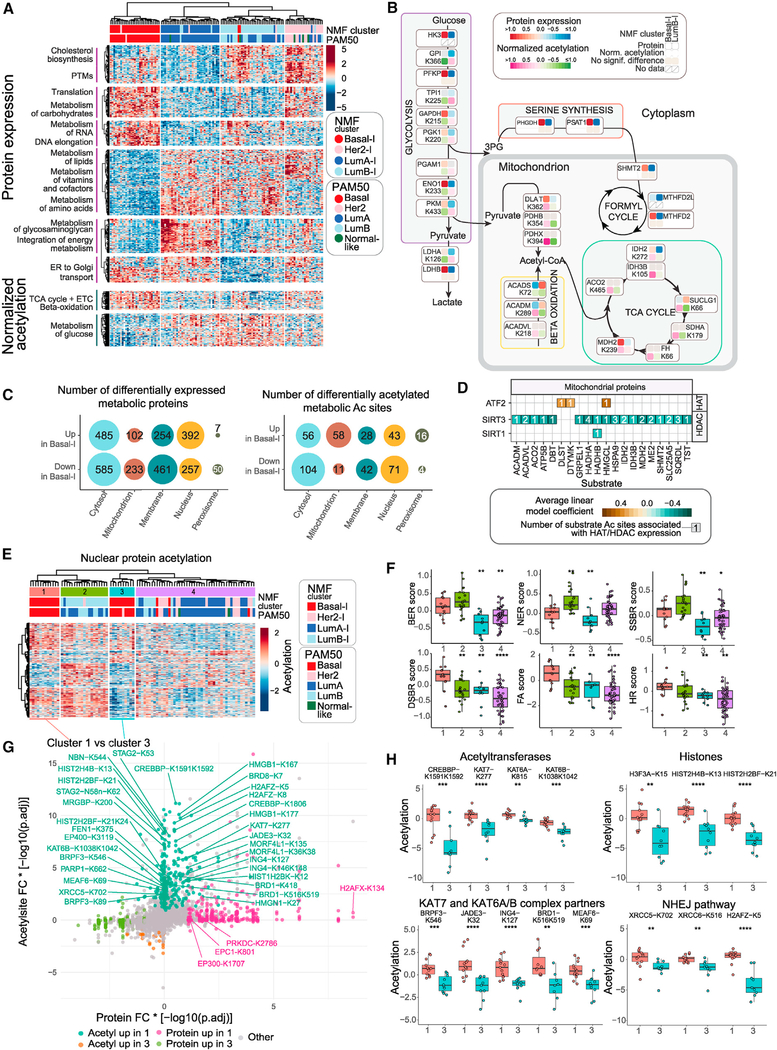
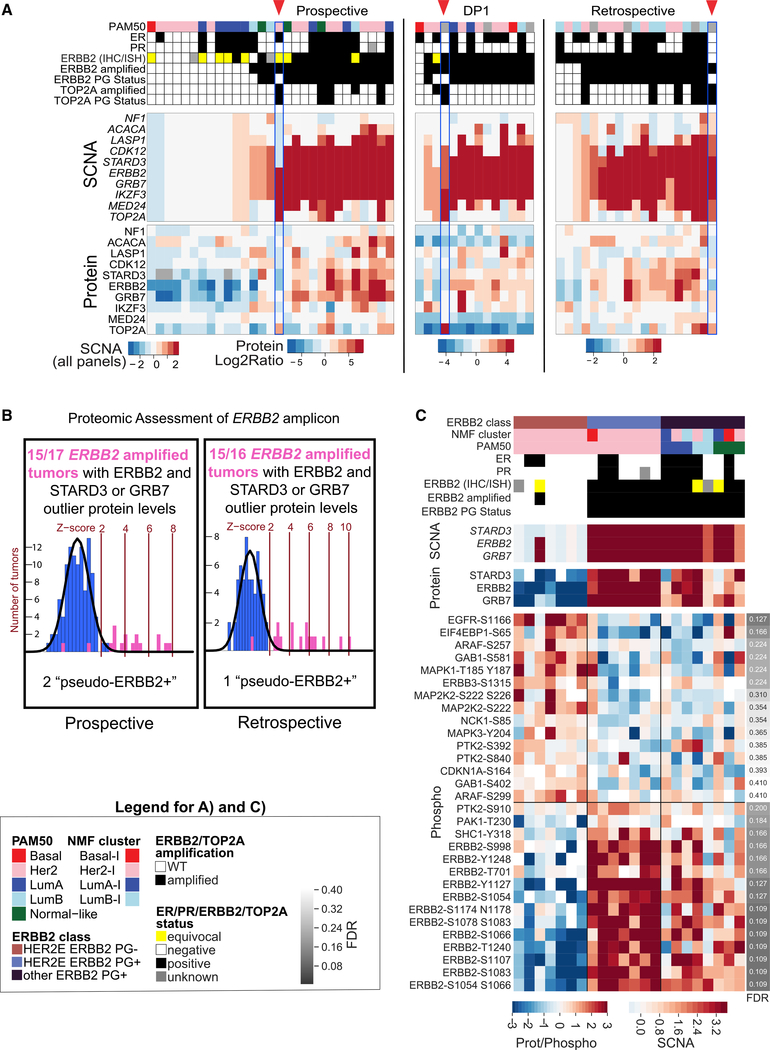
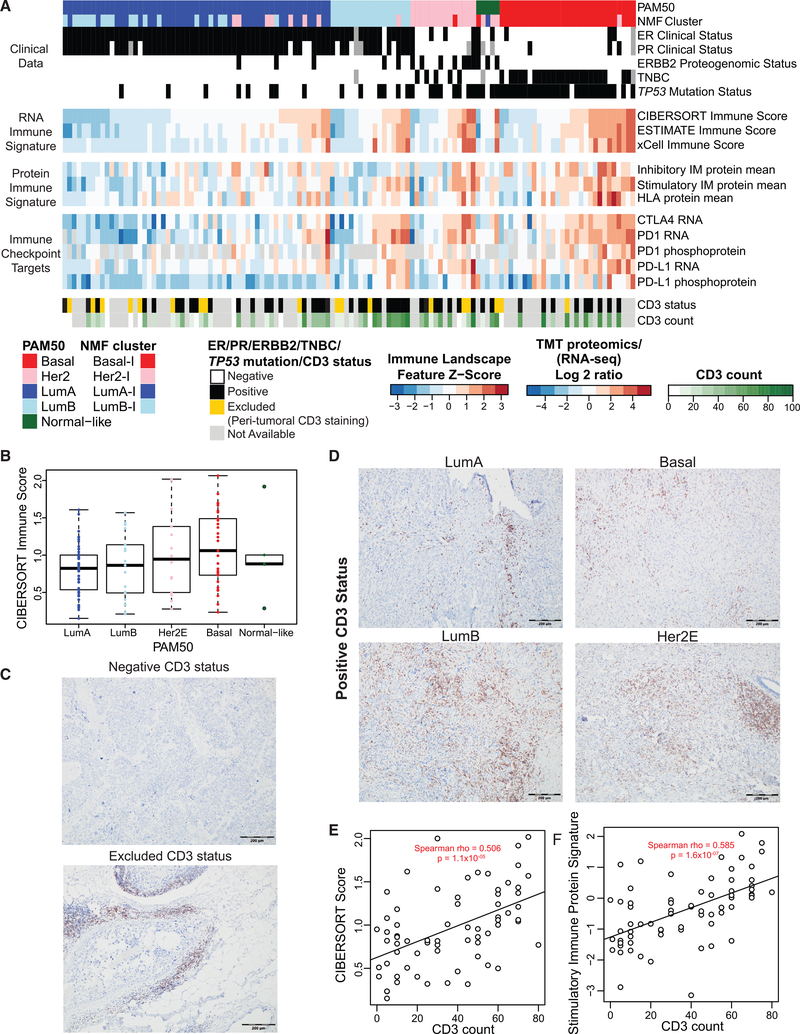
The integration of mass spectrometry-based proteomics with next-generation DNA and RNA sequencing profiles tumors more comprehensively. Here this "proteogenomics" approach was applied to 122 treatment-naive primary breast cancers accrued to preserve post-translational modifications, including protein phosphorylation and acetylation. Proteogenomics challenged standard breast cancer diagnoses, provided detailed analysis of the ERBB2 amplicon, defined tumor subsets that could benefit from immune checkpoint therapy, and allowed more accurate assessment of Rb status for prediction of CDK4/6 inhibitor responsiveness. Phosphoproteomics profiles uncovered novel associations between tumor suppressor loss and targetable kinases. Acetylproteome analysis highlighted acetylation on key nuclear proteins involved in the DNA damage response and revealed cross-talk between cytoplasmic and mitochondrial acetylation and metabolism. Our results underscore the potential of proteogenomics for clinical investigation of breast cancer through more accurate annotation of targetable pathways and biological features of this remarkably heterogeneous malignancy.
Keywords: CDK 4/6 inhibitors; CPTAC; acetylation; breast cancer; genomics; immune checkpoint therapy; mass spectrometry; phosphoproteomics; proteogenomics; proteomics.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests M.J.E reports ownership and royalties associated with Bioclassifier LLC through sales by Nanostring LLC and Veracyte for the “Prosigna” breast cancer prognostic test. He also reports ad hoc consulting for AstraZeneca, Foundation Medicine, G1 Therapeutics, Novartis, Sermonix, Abbvie, Lilly and Pfizer. B.Z. has received research funding from Bristol-Myers Squibb. S.A.C. is a scientific advisory board member of Kymera, PTM BioLabs, and Seer and ad hoc consultant to Pfizer and Biogen.
Figures
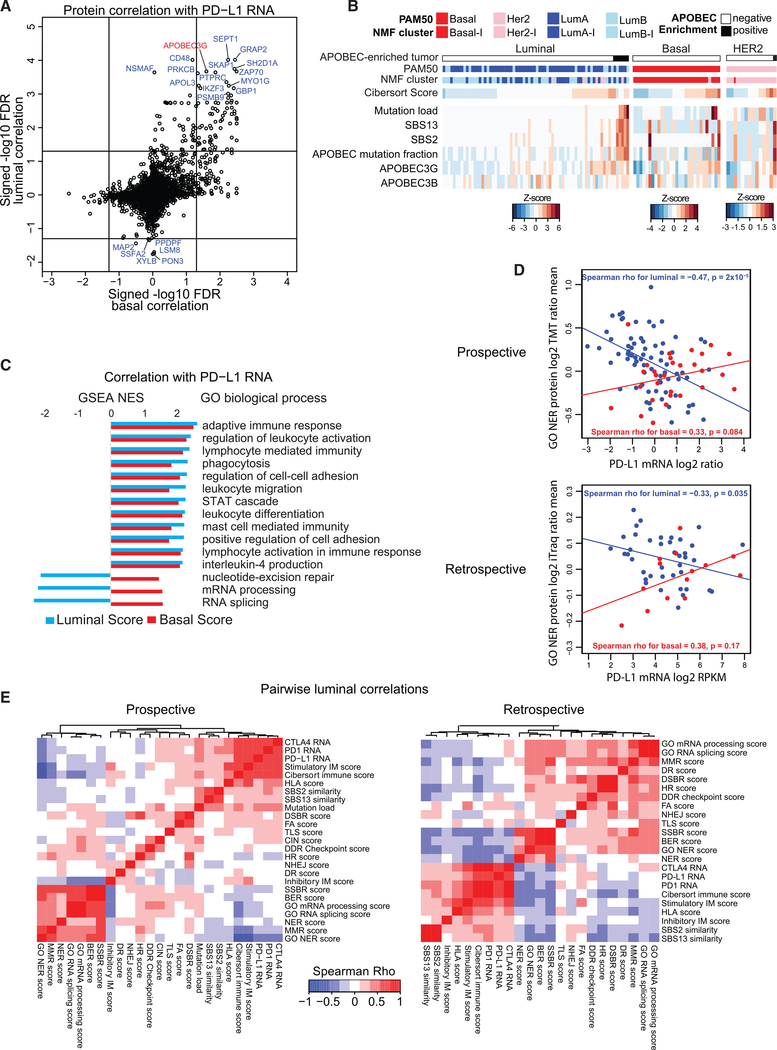
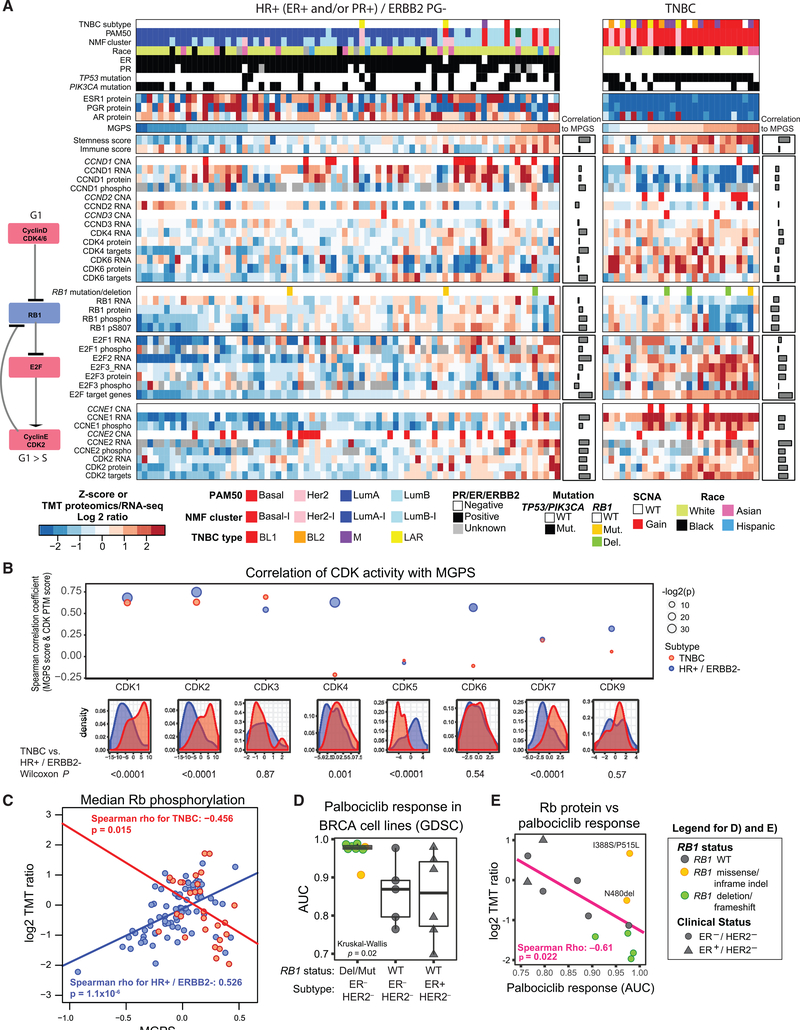
References
-
- Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, and Trajanoski Z (2015). Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 16, 64. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
