

Quantum Chemical Study Aimed at Modeling Efficient Aza-BODIPY NIR Dyes: Molecular and Electronic Structure, Absorption, and Emission Spectra
- PMID: 33212835
- PMCID: PMC7698449
- DOI: 10.3390/molecules25225361
Quantum Chemical Study Aimed at Modeling Efficient Aza-BODIPY NIR Dyes: Molecular and Electronic Structure, Absorption, and Emission Spectra
Abstract
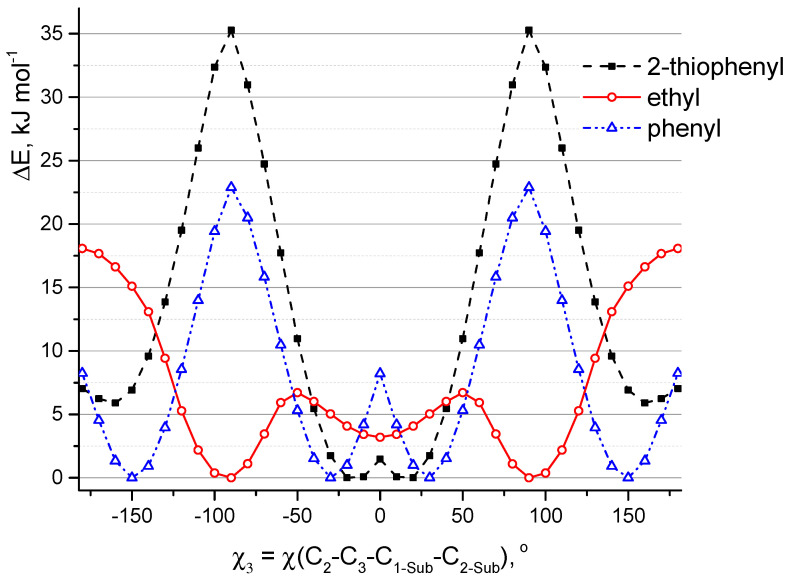
A comprehensive study of the molecular structure of aza-BODIPY and its derivatives, obtained by introduction of one or more substituents, was carried out. We considered the changes in the characteristics of the electronic and geometric structure of the unsubstituted aza-BODIPY introducing the following substituents into the dipyrrin core; phenyl, 2-thiophenyl, 2-furanyl, 3-pyridinyl, 4-pyridinyl, 2-pyridinyl, and ethyl groups. The ground-state geometries of the unsubstituted Aza-BODIPY and 27 derivatives were computed at the PBE/6-31G(d) and CAM-B3LYP/6-31+G(d,p) levels of theory. The time-dependent density-functional theory (TDDFT) together with FC vibronic couplings was used to investigate their absorption and emission spectra.
Keywords: absorption spectra; aza-BODIPY; intramolecular rotation; molecular structure; quantum chemical calculations; vibronic spectra.
Conflict of interest statement
The authors declare no conflict of interest.
Figures











References
-
- Ziessel R., Ulrich G., Harriman A. The chemistry of Bodipy: A new El Dorado for fluorescence tools. New J. Chem. 2007;31:496–501. doi: 10.1039/b617972j. - DOI
-
- Vodyanova O.S., Kochergin B.A., Usoltsev S.D., Marfin Y.S., Rumyantsev E.V., Aleksakhina E.L., Tomilova I.K. BODIPY dyes in bio environment: Spectral characteristics and possibilities for practical application. J. Photochem. Photobiol. A Chem. 2018;350:44–51. doi: 10.1016/j.jphotochem.2017.09.049. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
