

Nicotine Modulates MyD88-Dependent Signaling Pathway in Macrophages during Mycobacterial Infection
- PMID: 33212859
- PMCID: PMC7698335
- DOI: 10.3390/microorganisms8111804
Nicotine Modulates MyD88-Dependent Signaling Pathway in Macrophages during Mycobacterial Infection
Abstract
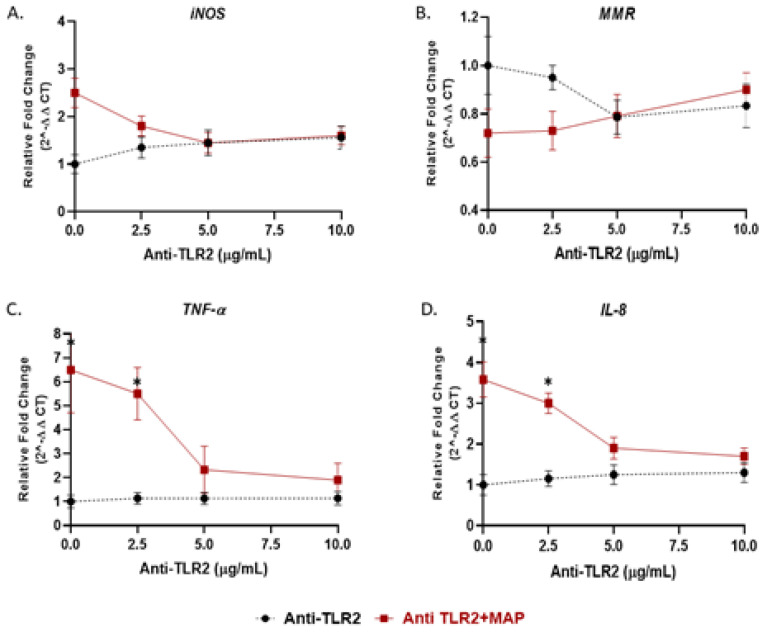
Recently, we reported that cigarette smoking, and especially nicotine, increases susceptibility to mycobacterial infection and exacerbates inflammation in patients with Crohn's disease (CD). The macrophagic response to Mycobacterium avium subspecies paratuberculosis (MAP) in CD and Mycobacteria tuberculosis (MTB) continues to be under investigation. The role of toll-like-receptors (TLRs) and cytoplasmic adaptor protein (MyD88) in proinflammatory response during Mycobacterial infection has been suggested. However, the mechanism of how nicotine modulates macrophage response during infection in CD and exacerbates inflammatory response remain unclear. In this study, we elucidated the mechanistic role of nicotine in modulating MyD88-dependent/TLR pathway signaling in a macrophage system during mycobacterial infection. The data demonstrated that MAP infection in THP-1 derived macrophages was mediated through TLR2 and MyD88 leading to increase in IL-8 in expression and production. On the other hand, LPS-representing, Gram-negative bacteria mediated macrophage response through TLR4. Blocking TLR2 and TLR4 with antagonists voided the effect of MAP, and LPS, respectively in macrophages and reversed response with decrease in expression of iNOS, TNF-α and IL-8. Interestingly, nicotine in infected macrophages significantly (1) downregulated TLR2 and TLR4 expression, (2) activated MyD88, (3) increased M1/M2 ratio, and (4) increased expression and secretion of proinflammatory cytokines especially IL-8, as seen in CD smokers. We also discovered that blocking macrophages during MAP infection with MyD88 antagonist significantly decreased response which illustrates the key role for MyD88 during infection. Surprisingly, dual treatment of MAP-infected macrophages with MyD88 antagonist and nicotine absolutely impaired immune response and decreased MAP viability, which clearly validate the inflammatory role of nicotine in macrophages through TLR2/MyD88 pathway during infection. This is the first report to describe the mechanism by which nicotine modulates TLR2/MyDD88 and exacerbates inflammation in CD smokers associated with infection.
Keywords: Crohn’s disease; MAP; Macrophages; MyD88; Nicotine; TLR2.
Conflict of interest statement
Authors declare no conflict of interest.
Figures






Similar articles
-
Mystery Solved: Why Smoke Extract Worsens Disease in Smokers with Crohn's Disease and Not Ulcerative Colitis? Gut MAP!Microorganisms. 2020 May 2;8(5):666. doi: 10.3390/microorganisms8050666. Microorganisms. 2020. PMID: 32370298 Free PMC article.
-
Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals.J Immunol. 2003 Nov 1;171(9):4758-64. doi: 10.4049/jimmunol.171.9.4758. J Immunol. 2003. PMID: 14568952
-
Glycopeptidolipids from Mycobacterium avium promote macrophage activation in a TLR2- and MyD88-dependent manner.J Leukoc Biol. 2006 Aug;80(2):415-23. doi: 10.1189/jlb.1205702. Epub 2006 Jun 7. J Leukoc Biol. 2006. PMID: 16760377
-
Divergent Effect of Cigarette Smoke on Innate Immunity in Inflammatory Bowel Disease: A Nicotine-Infection Interaction.Int J Mol Sci. 2020 Aug 13;21(16):5801. doi: 10.3390/ijms21165801. Int J Mol Sci. 2020. PMID: 32823518 Free PMC article. Review.
-
Effect of Nicotine on Immune System Function.Adv Pharm Bull. 2023 Jan;13(1):69-78. doi: 10.34172/apb.2023.008. Epub 2022 Jan 4. Adv Pharm Bull. 2023. PMID: 36721811 Free PMC article. Review.
Cited by
-
Nicotine in Inflammatory Diseases: Anti-Inflammatory and Pro-Inflammatory Effects.Front Immunol. 2022 Feb 18;13:826889. doi: 10.3389/fimmu.2022.826889. eCollection 2022. Front Immunol. 2022. PMID: 35251010 Free PMC article. Review.
-
Nicotine Increases Macrophage Survival through α7nAChR/NF-κB Pathway in Mycobacterium avium paratuberculosis Infection.Microorganisms. 2021 May 18;9(5):1086. doi: 10.3390/microorganisms9051086. Microorganisms. 2021. PMID: 34070119 Free PMC article.
-
Lactobacillus murinus alleviate intestinal ischemia/reperfusion injury through promoting the release of interleukin-10 from M2 macrophages via Toll-like receptor 2 signaling.Microbiome. 2022 Mar 3;10(1):38. doi: 10.1186/s40168-022-01227-w. Microbiome. 2022. PMID: 35241180 Free PMC article.
-
MCT4 inhibition attenuates inflammatory response to Mycobacterium avium paratuberculosis infection and restores intestinal epithelial integrity in vitro.Front Immunol. 2025 Apr 14;16:1562100. doi: 10.3389/fimmu.2025.1562100. eCollection 2025. Front Immunol. 2025. PMID: 40297589 Free PMC article.
-
Effect of Nicotine on Pulmonary Pathogenic Bacteria.Curr Microbiol. 2024 Nov 8;81(12):450. doi: 10.1007/s00284-024-03977-2. Curr Microbiol. 2024. PMID: 39514085 Review.
References
-
- Nazareth N., Magro F., Machado E., Ribeiro T.G., Martinho A., Rodrigues P., Abreu C. Prevalence of Mycobacteriumavium subsp. paratuberculosis and Escherichia coli in blood samples from patients with inflammatory bowel disease. Med. Microbiol. Immunol. 2015;204:681–692. doi: 10.1007/s00430-015-0420-3. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
