

Endoplasmic reticulum stress signals in the tumour and its microenvironment
- PMID: 33214692
- PMCID: PMC7927882
- DOI: 10.1038/s41568-020-00312-2
Endoplasmic reticulum stress signals in the tumour and its microenvironment
Abstract
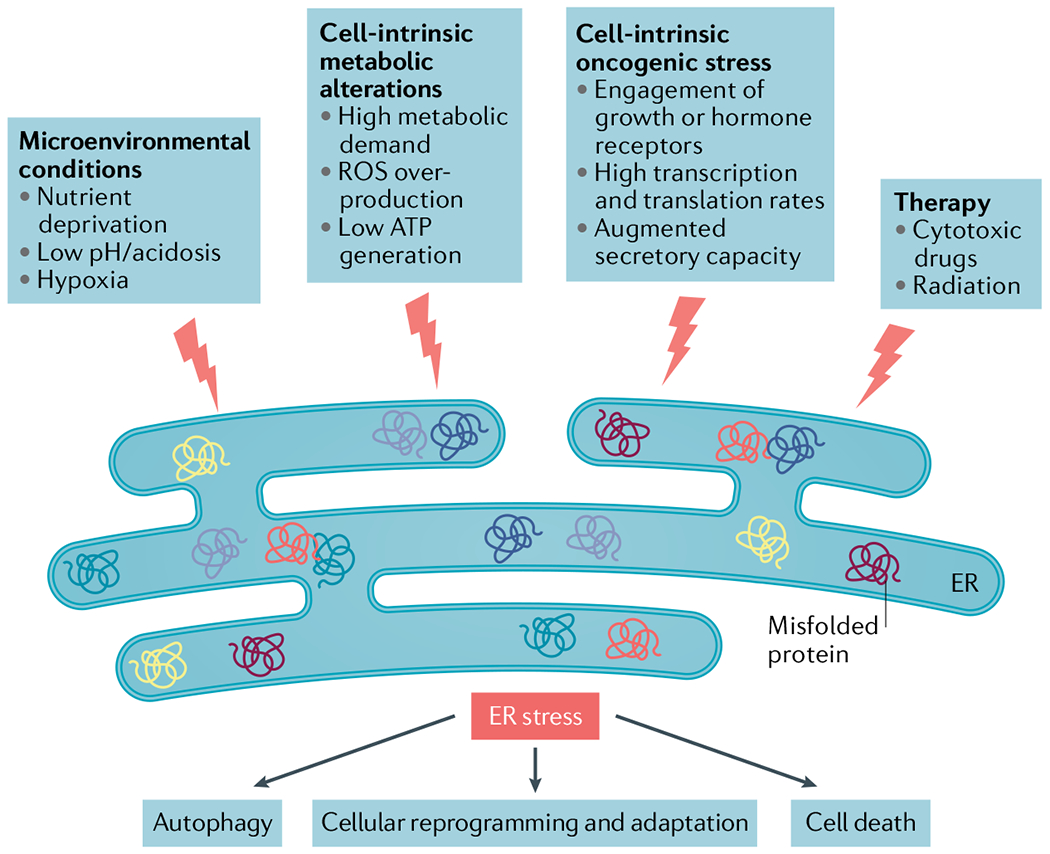
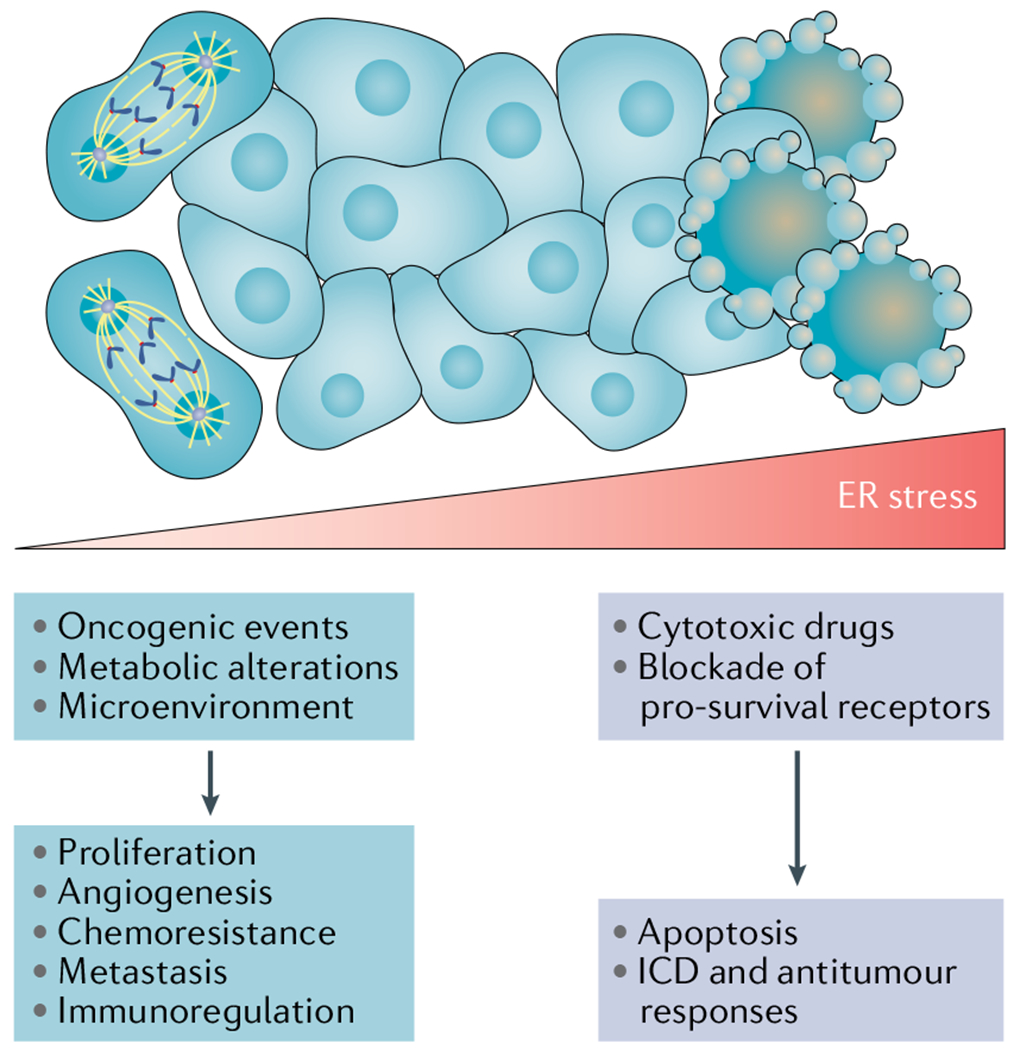
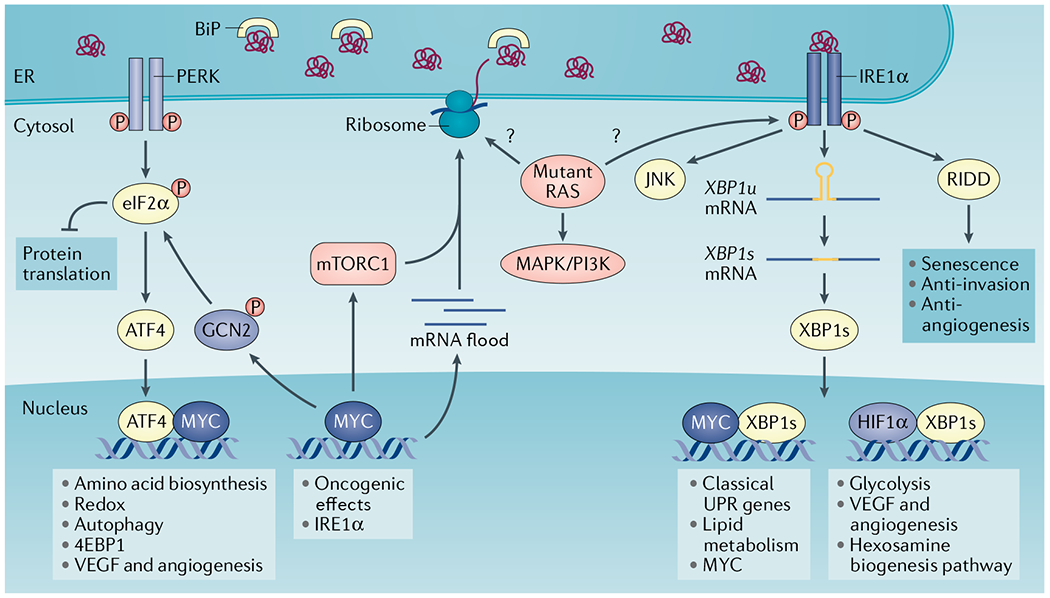
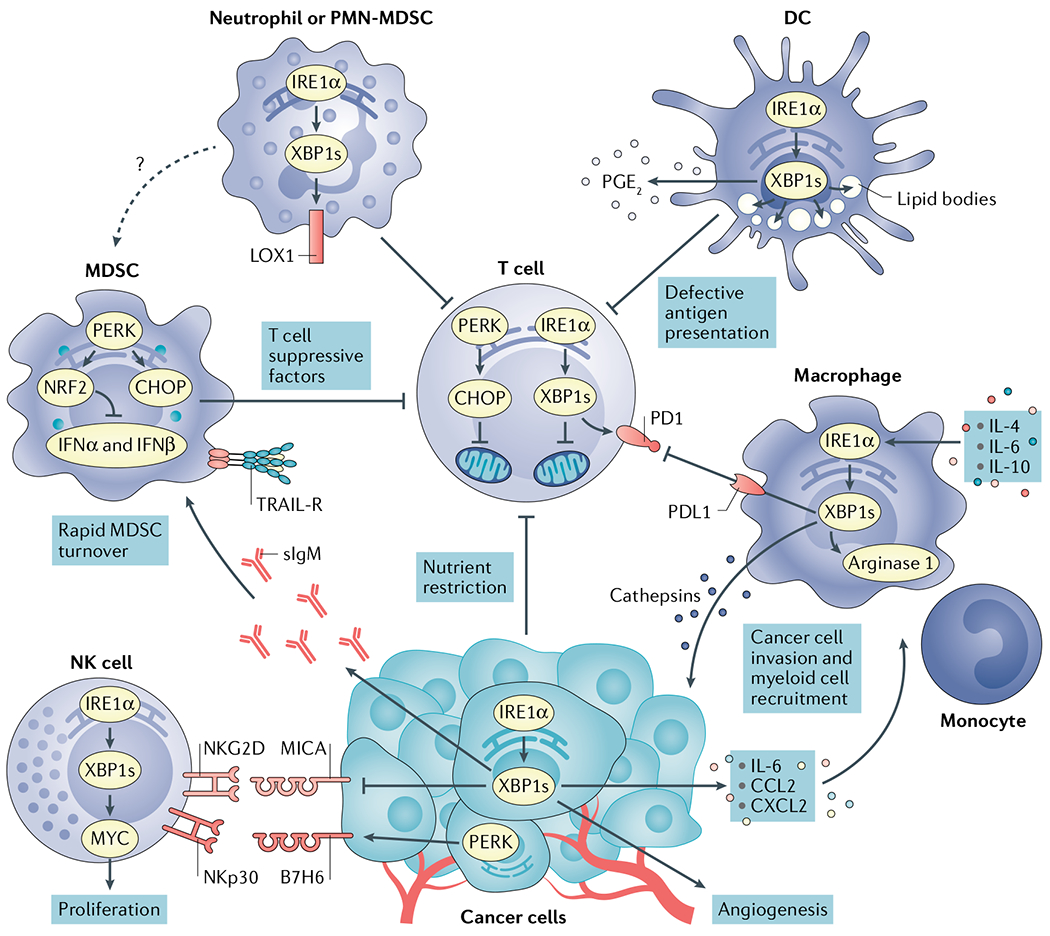
Protein handling, modification and folding in the endoplasmic reticulum (ER) are tightly regulated processes that determine cell function, fate and survival. In several tumour types, diverse oncogenic, transcriptional and metabolic abnormalities cooperate to generate hostile microenvironments that disrupt ER homeostasis in malignant and stromal cells, as well as infiltrating leukocytes. These changes provoke a state of persistent ER stress that has been demonstrated to govern multiple pro-tumoural attributes in the cancer cell while dynamically reprogramming the function of innate and adaptive immune cells. Aberrant activation of ER stress sensors and their downstream signalling pathways have therefore emerged as key regulators of tumour growth and metastasis as well as response to chemotherapy, targeted therapies and immunotherapy. In this Review, we discuss the physiological inducers of ER stress in the tumour milieu, the interplay between oncogenic signalling and ER stress response pathways in the cancer cell and the profound immunomodulatory effects of sustained ER stress responses in tumours.
Conflict of interest statement
Competing interests
X.C. and J.R.C.-R. hold patents on the use of ER stress and UPR modulators for cancer therapy. X.C. reports research funding from Fosun Pharma.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
