

Congenital myasthenic syndrome due to a TOR1AIP1 mutation: a new disease pathway for impaired synaptic transmission
- PMID: 33215087
- PMCID: PMC7660151
- DOI: 10.1093/braincomms/fcaa174
Congenital myasthenic syndrome due to a TOR1AIP1 mutation: a new disease pathway for impaired synaptic transmission
Abstract
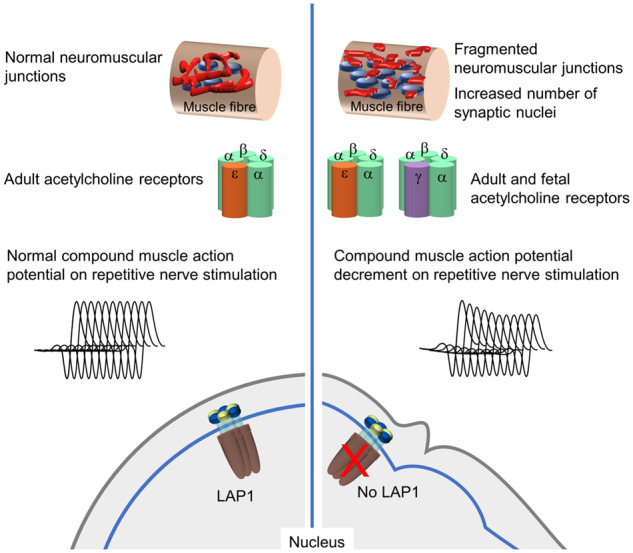
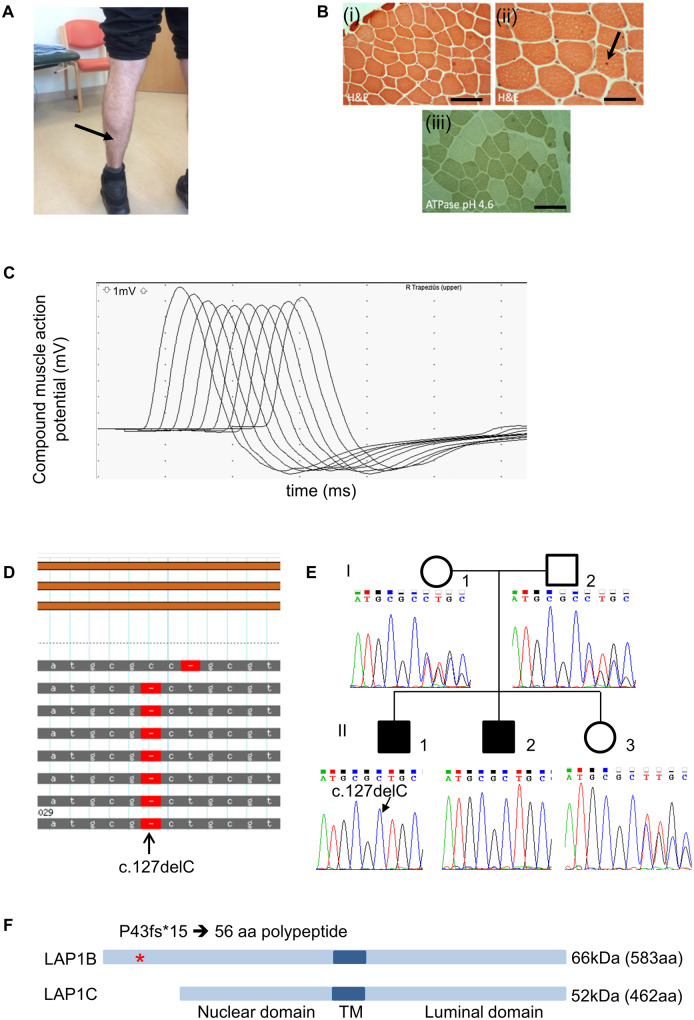
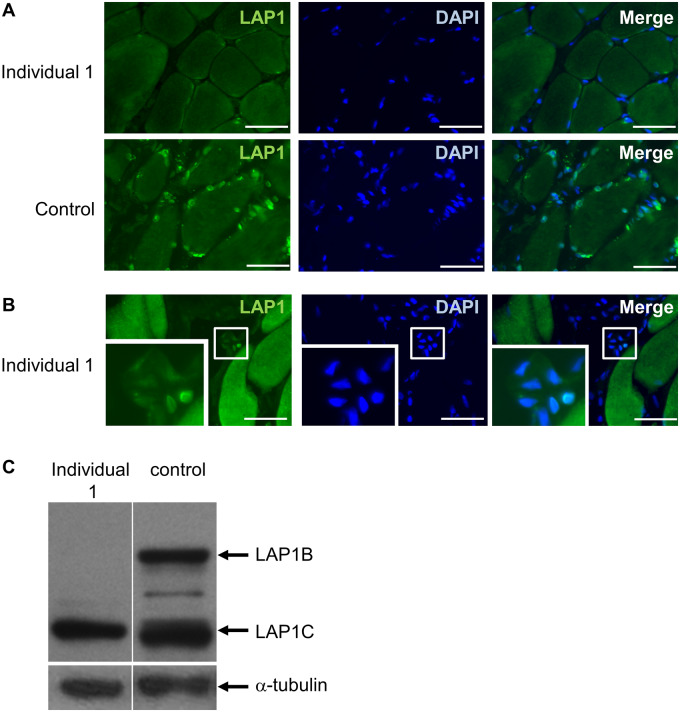
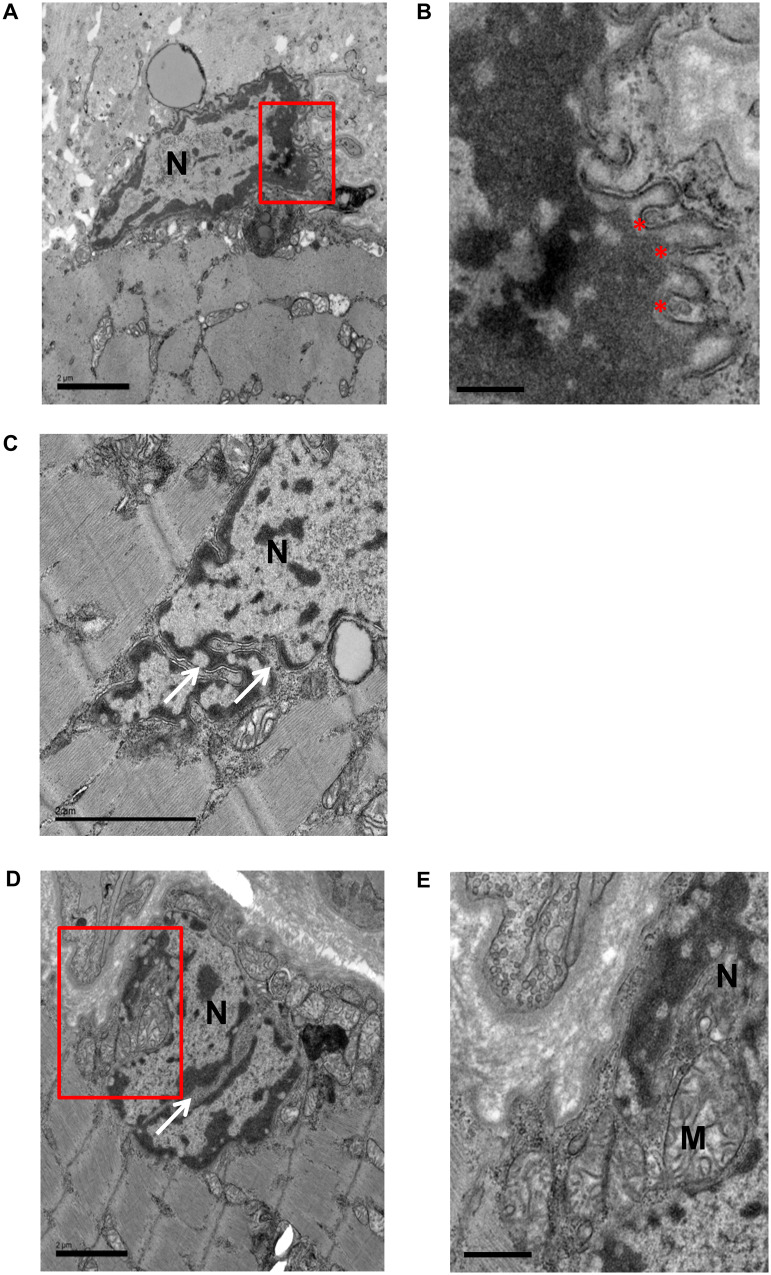
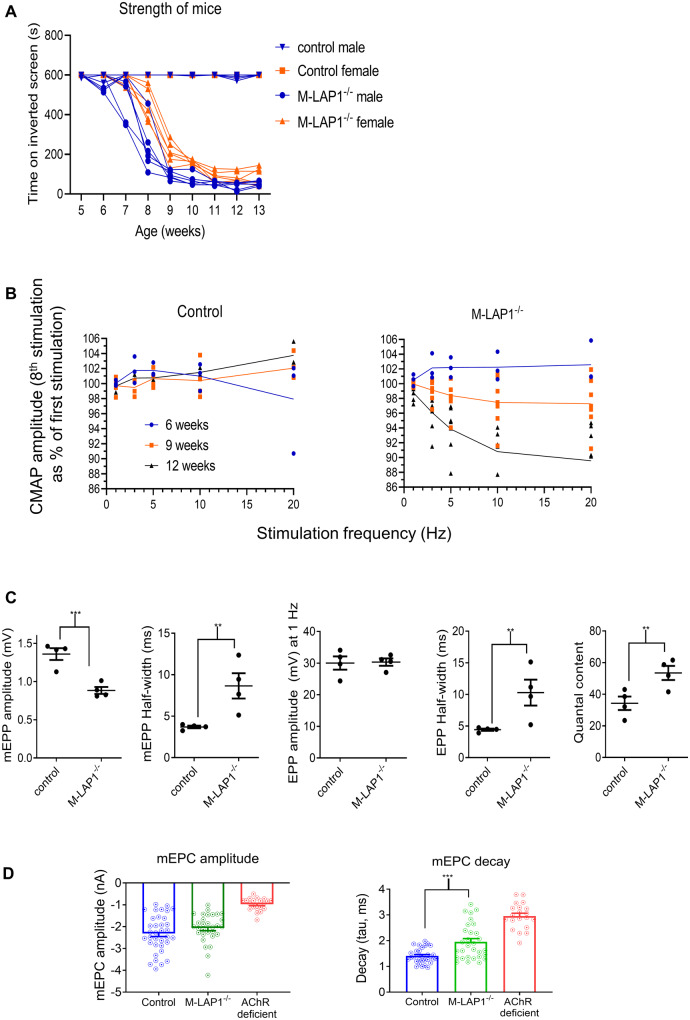
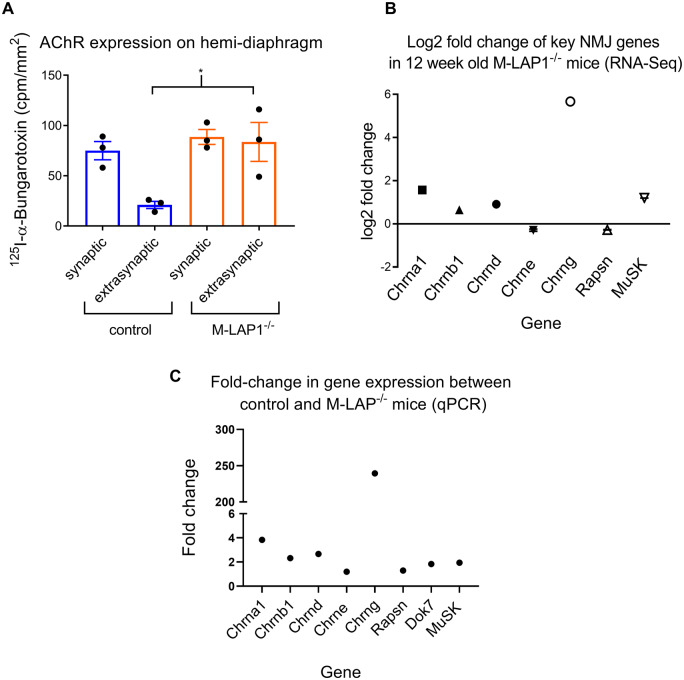
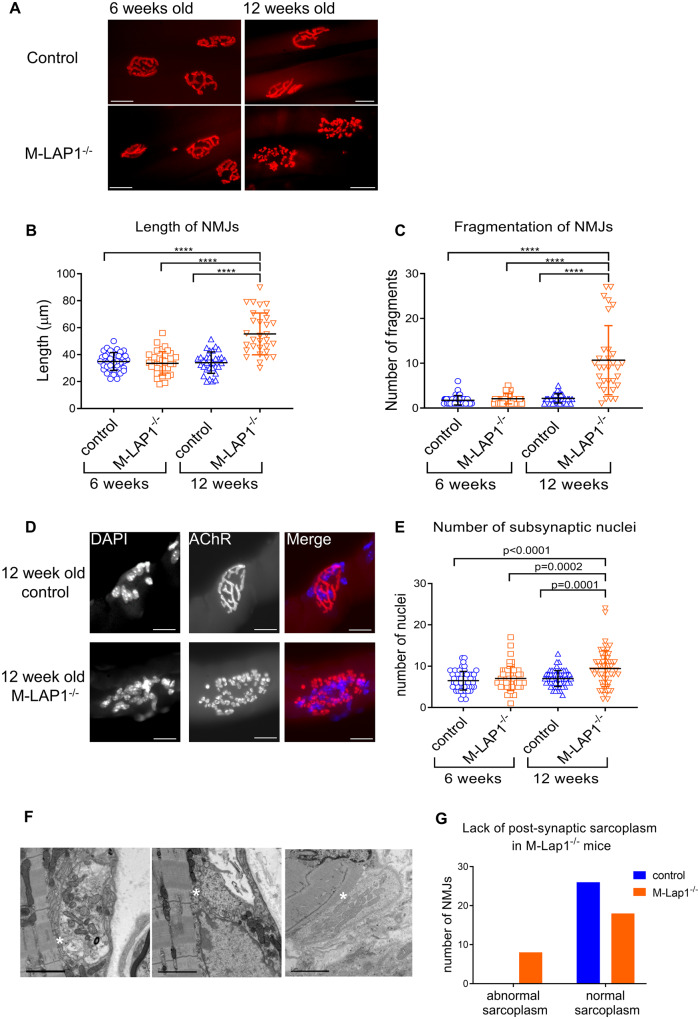
Congenital myasthenic syndromes are inherited disorders characterized by fatiguable muscle weakness resulting from impaired signal transmission at the neuromuscular junction. Causative mutations have been identified in genes that can affect the synaptic function or structure. We identified a homozygous frameshift deletion c.127delC, p. Pro43fs in TOR1AIP1 in two siblings with limb-girdle weakness and impaired transmission at the neuromuscular synapse. TOR1AIP1 encodes the inner nuclear membrane protein lamin-associated protein 1. On muscle biopsy from the index case, lamin-associated protein 1 was absent from myonuclei. A mouse model with lamin-associated protein 1 conditionally knocked out in striated muscle was used to analyse the role of lamin-associated protein 1 in synaptic dysfunction. Model mice develop fatiguable muscle weakness as demonstrated by using an inverted screen hang test. Electromyography on the mice revealed a decrement on repetitive nerve stimulation. Ex vivo analysis of hemi-diaphragm preparations showed both miniature and evoked end-plate potential half-widths were prolonged which was associated with upregulation of the foetal acetylcholine receptor γ subunit. Neuromuscular junctions on extensor digitorum longus muscles were enlarged and fragmented, and the number of subsynaptic nuclei was significantly increased. Following these findings, electromyography was performed on cases of other nuclear envelopathies caused by mutations in LaminA/C or emerin, but decrement on repetitive nerve stimulation or other indications of defective neuromuscular transmission were not seen. Thus, this report highlights the first nuclear membrane protein in which defective function can lead to impaired synaptic transmission.
Keywords: congenital myasthenic syndromes; envelopathy; myasthenia; neuromuscular junction; nuclear envelope protein.
© The Author(s) (2020). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Similar articles
-
Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with 'limb-girdle myasthenia'.Brain. 2006 Aug;129(Pt 8):2061-76. doi: 10.1093/brain/awl200. Brain. 2006. PMID: 16870884
-
A TOR1AIP1 variant segregating with an early onset limb girdle myasthenia-Support for the role of LAP1 in NMJ function and disease.Neuropathol Appl Neurobiol. 2022 Feb;48(1):e12743. doi: 10.1111/nan.12743. Epub 2021 Jul 13. Neuropathol Appl Neurobiol. 2022. PMID: 34164833
-
Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy.Brain. 2014 Sep;137(Pt 9):2429-43. doi: 10.1093/brain/awu160. Epub 2014 Jun 20. Brain. 2014. PMID: 24951643
-
Congenital Myasthenic Syndromes.Neurol Clin. 2018 May;36(2):367-378. doi: 10.1016/j.ncl.2018.01.007. Neurol Clin. 2018. PMID: 29655455 Review.
-
Effective Treatment With Albuterol in DOK7 Congenital Myasthenic Syndrome in Children.Pediatr Neurol. 2016 Jan;54:85-7. doi: 10.1016/j.pediatrneurol.2015.09.019. Epub 2015 Nov 6. Pediatr Neurol. 2016. PMID: 26552645 Review.
Cited by
-
Preimplantation genetic testing as a means of preventing hereditary congenital myasthenic syndrome caused by RAPSN.Mol Genet Genomic Med. 2024 Mar;12(3):e2409. doi: 10.1002/mgg3.2409. Mol Genet Genomic Med. 2024. PMID: 38511267 Free PMC article.
-
LAP1 Interactome Profiling Provides New Insights into LAP1's Physiological Functions.Int J Mol Sci. 2024 Dec 10;25(24):13235. doi: 10.3390/ijms252413235. Int J Mol Sci. 2024. PMID: 39769001 Free PMC article.
-
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review.Int J Mol Sci. 2023 Feb 13;24(4):3730. doi: 10.3390/ijms24043730. Int J Mol Sci. 2023. PMID: 36835142 Free PMC article. Review.
-
Congenital myasthenic syndromes in adults: clinical features, diagnosis and long-term prognosis.Brain. 2024 Nov 4;147(11):3849-3862. doi: 10.1093/brain/awae124. Brain. 2024. PMID: 38696726 Free PMC article.
-
Congenital myasthenic syndromes: increasingly complex.Curr Opin Neurol. 2024 Oct 1;37(5):493-501. doi: 10.1097/WCO.0000000000001300. Epub 2024 Jul 25. Curr Opin Neurol. 2024. PMID: 39051439 Free PMC article. Review.
References
-
- Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 1994; 8: 323–7. - PubMed
-
- Carlson CG, Roshek DM. Adult dystrophic (mdx) endplates exhibit reduced quantal size and enhanced quantal variation. Pflügers Arch - Eur J Physiol 2001; 442: 369–75. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials