

PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells
- PMID: 33218077
- PMCID: PMC7698885
- DOI: 10.3390/ijms21228711
PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells
Abstract
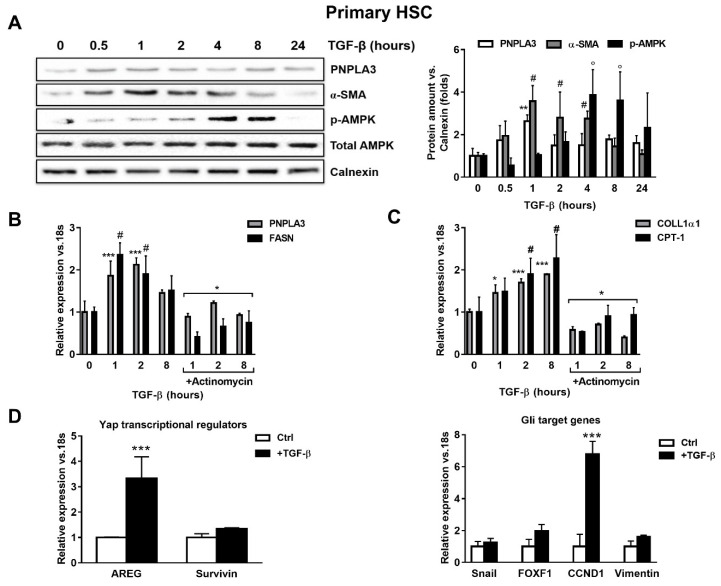
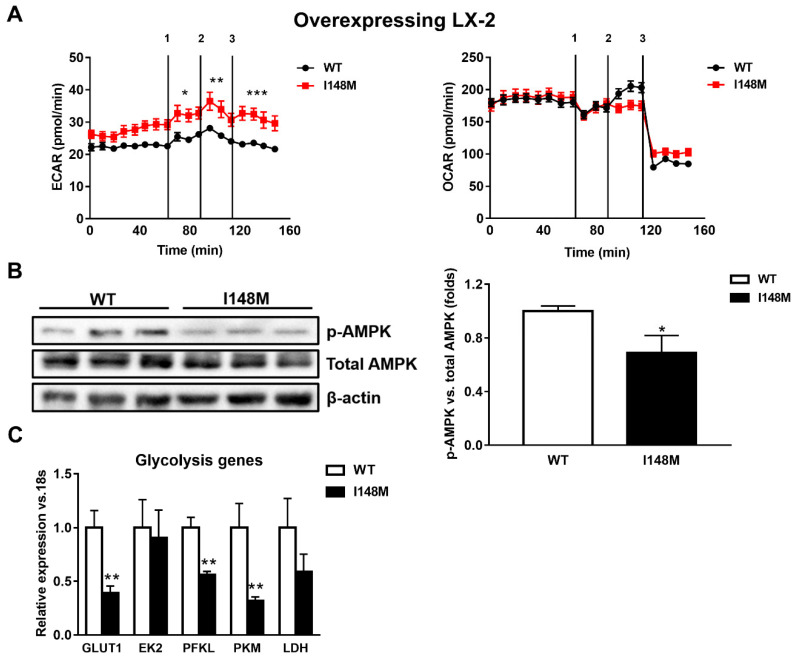
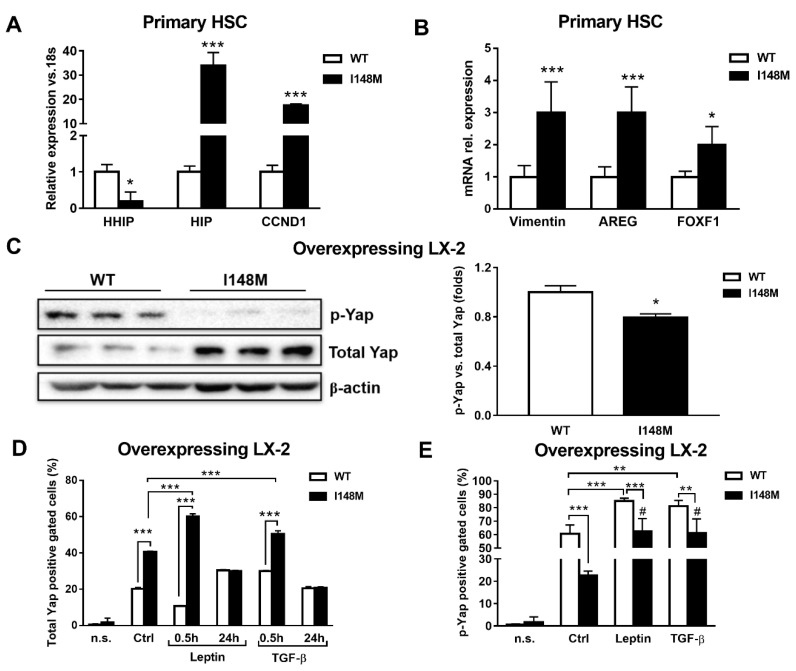
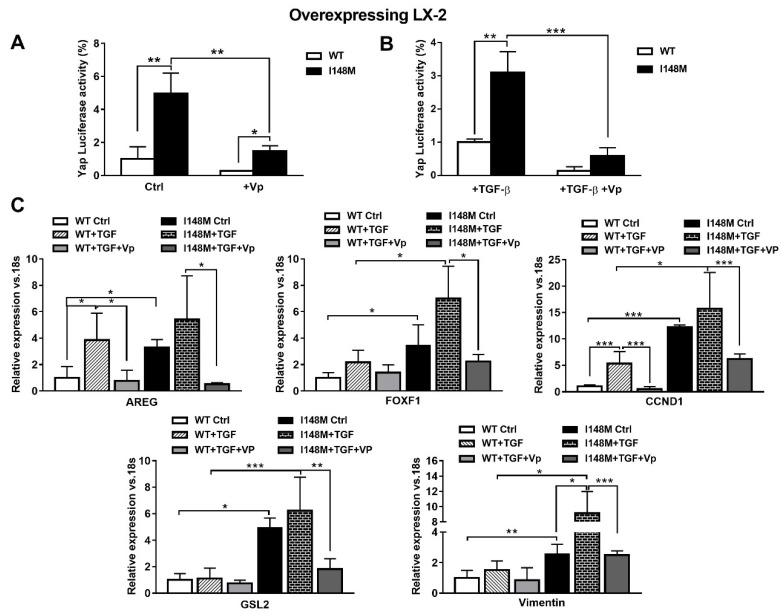
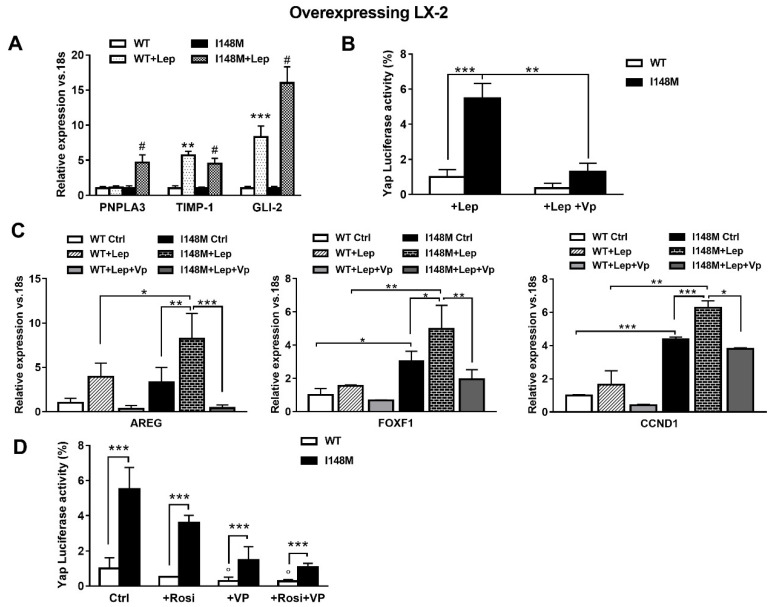
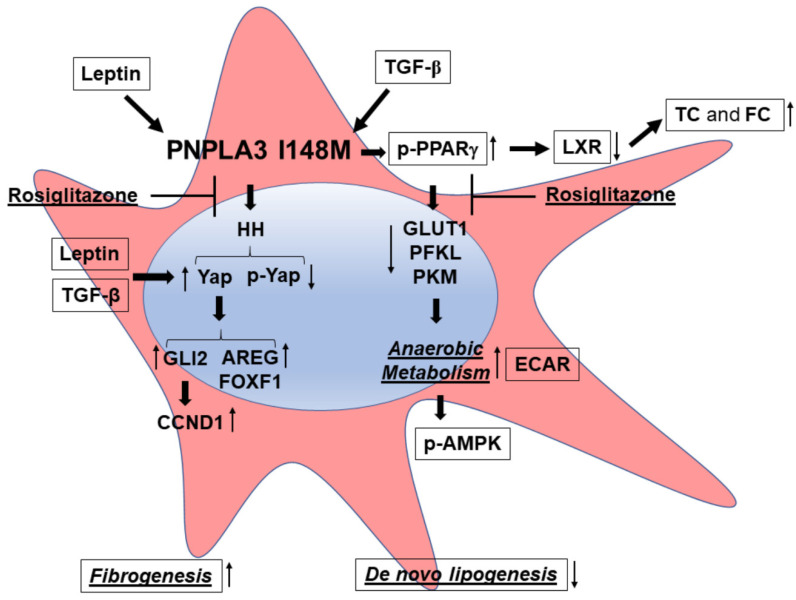
Liver fibrosis represents the wound healing response to sustained hepatic injury with activation of hepatic stellate cells (HSCs). The I148M variant of the PNPLA3 gene represents a risk factor for development of severe liver fibrosis. Activated HSCs carrying the I148M variant display exacerbated pro-inflammatory and pro-fibrogenic features. We aimed to examine whether the I148M variant may impair Hedgehog and Yap signaling, as key pathways implicated in the control of energy expenditure and maintenance of myofibroblastic traits. First, we show that TGF-β rapidly up-regulated the PNPLA3 transcript and protein and Yap/Hedgehog target gene expression. In addition, HSCs overexpressing PNPLA3 I148M boosted anaerobic glycolysis, as supported by higher lactate release and decreased phosphorylation of the energy sensor AMPK. These cells displayed higher Yap and Hedgehog signaling, due to accumulation of total Yap protein, Yap promoter activity and increased downstream targets expression, compared to WT cells. HSCs exposed to TGF-β and leptin rapidly increased total Yap, together with a reduction in its inhibited form, phosphorylated Yap. In line, Yap-specific inhibitor Verteporfin strongly abolished Yap-mediated genes expression, at baseline as well as after TGF-β and leptin treatments in HSCs with I148M PNPLA3. Finally, Yap transcriptional activity was strongly reduced by a combination of Verteporfin and Rosiglitazone, a PPARγ synthetic agonist. In conclusion, HSCs carrying the PNPLA3 variant show activated Yap/Hedgehog pathways, resulting in altered anaerobic glycolysis and enhanced synthesis of Hedgehog markers and sustained Yap signaling. TGF-β and leptin exacerbate Yap/Hedgehog-related fibrogenic genes expression, while Yap inhibitors and PPARγ agonists abrogate these effects in PNPLA3 I148M carrying HSCs.
Keywords: cell metabolism; genetic polymorphism; intracellular signaling; non-alcoholic fatty liver disease.
Conflict of interest statement
Michael Trauner received speaker fees from Bristol-Myers Squibb (BMS), Falk Foundation, Gilead, Intercept and Merck Sharp & Dohme (MSD); advisory board fees from Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, GENFIT, Gilead, Intercept, Jannsen, MSD, Novartis, Phenex, and Regulus; travel grants from AbbVie, Falk, Gilead and Intercept; and unrestricted research grants from Albireo, CymaBay, Falk, Gilead, Intercept, MSD and Takeda. He is also coinventor of patents on the medical use of norUDCA filed by the Medical University of Graz. All the other authors declare no conflicts of interest.
Figures
References
-
- Angulo P., Kleiner D.E., Dam-Larsen S., Adams L.A., Bjornsson E.S., Charatcharoenwitthaya P., Mills P.R., Keach J.C., Lafferty H.D., Stahler A., et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149:389–397. doi: 10.1053/j.gastro.2015.04.043. - DOI - PMC - PubMed
-
- Vilar-Gomez E., Calzadilla-Bertot L., Wong V.W.-S., Castellanos M., La Fuente R.A.-D., Metwally M., Eslam M., Gonzalez-Fabian L., Sanz M.A.-Q., Conde-Martin A.F., et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology. 2018;155:443–457. doi: 10.1053/j.gastro.2018.04.034. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
