

The essential elements of Alzheimer's disease
- PMID: 33219130
- PMCID: PMC7948403
- DOI: 10.1074/jbc.REV120.008207
The essential elements of Alzheimer's disease
Abstract
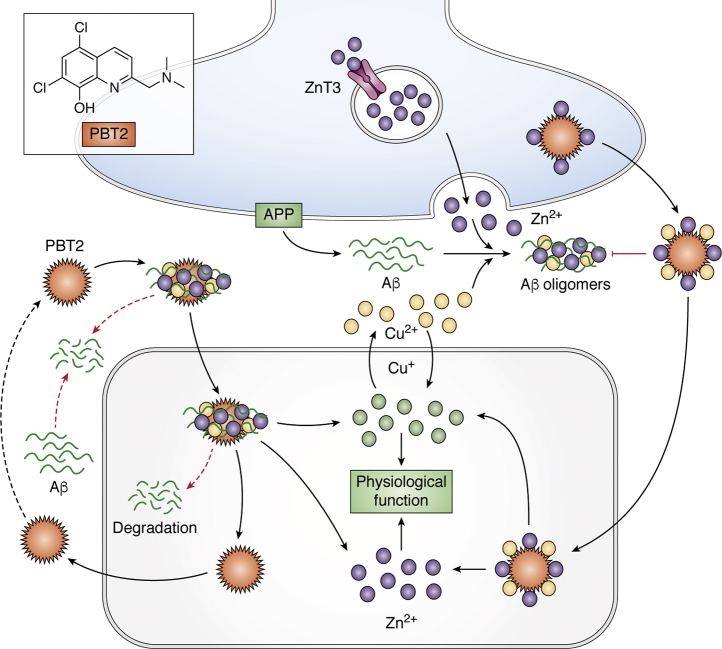
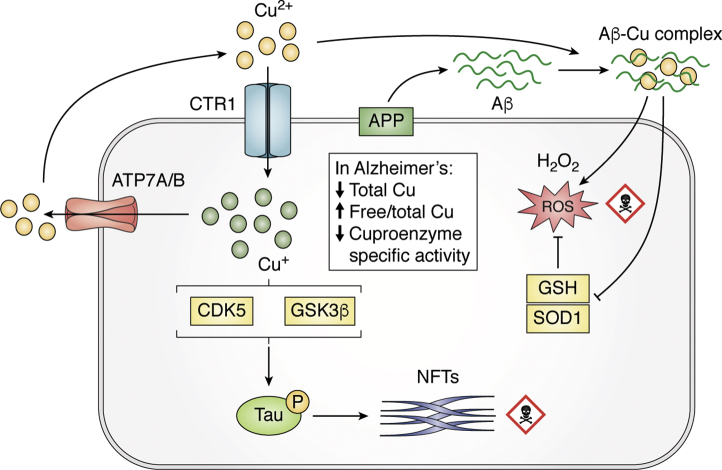
Treatments for Alzheimer's disease (AD) directed against the prominent amyloid plaque neuropathology are yet to be proved effective despite many phase 3 clinical trials. There are several other neurochemical abnormalities that occur in the AD brain that warrant renewed emphasis as potential therapeutic targets for this disease. Among those are the elementomic signatures of iron, copper, zinc, and selenium. Here, we review these essential elements of AD for their broad potential to contribute to Alzheimer's pathophysiology, and we also highlight more recent attempts to translate these findings into therapeutics. A reinspection of large bodies of discovery in the AD field, such as this, may inspire new thinking about pathogenesis and therapeutic targets.
Keywords: Alzheimer’s disease; PBT2; clioquinol; copper; ferroptosis; iron; selenium; zinc.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest A. I. B. is a shareholder in Alterity Therapeutics Ltd, Cogstate Ltd, and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
