

The Role of Cholesterol in α-Synuclein and Lewy Body Pathology in GBA1 Parkinson's Disease
- PMID: 33219714
- PMCID: PMC8247417
- DOI: 10.1002/mds.28396
The Role of Cholesterol in α-Synuclein and Lewy Body Pathology in GBA1 Parkinson's Disease
Abstract
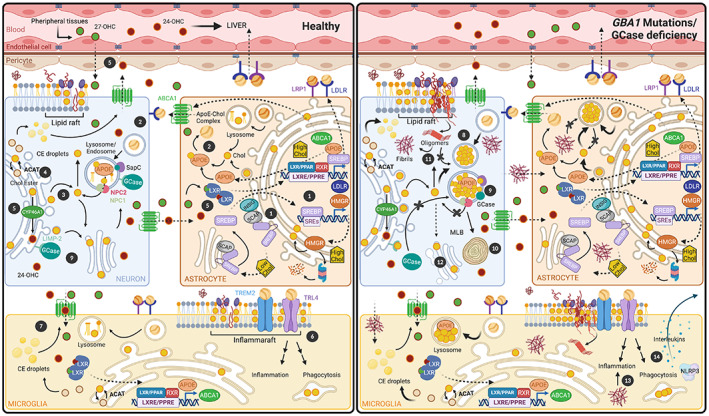
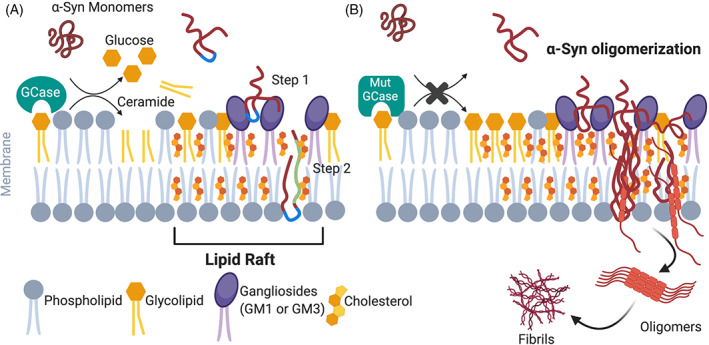
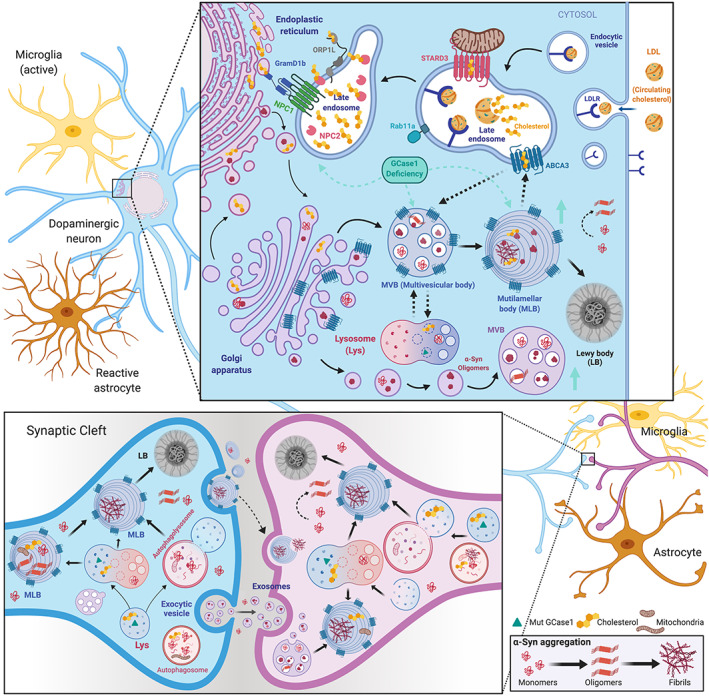
Parkinson's disease (PD) is a progressive neurodegenerative disease where dopaminergic neurons in the substantia nigra are lost, resulting in a decrease in striatal dopamine and, consequently, motor control. Dopaminergic degeneration is associated with the appearance of Lewy bodies, which contain membrane structures and proteins, including α-synuclein (α-Syn), in surviving neurons. PD displays a multifactorial pathology and develops from interactions between multiple elements, such as age, environmental conditions, and genetics. Mutations in the GBA1 gene represent one of the major genetic risk factors for PD. This gene encodes an essential lysosomal enzyme called β-glucocerebrosidase (GCase), which is responsible for degrading the glycolipid glucocerebroside into glucose and ceramide. GCase can generate glucosylated cholesterol via transglucosylation and can also degrade the sterol glucoside. Although the molecular mechanisms that predispose an individual to neurodegeneration remain unknown, the role of cholesterol in PD pathology deserves consideration. Disturbed cellular cholesterol metabolism, as reflected by accumulation of lysosomal cholesterol in GBA1-associated PD cellular models, could contribute to changes in lipid rafts, which are necessary for synaptic localization and vesicle cycling and modulation of synaptic integrity. α-Syn has been implicated in the regulation of neuronal cholesterol, and cholesterol facilitates interactions between α-Syn oligomers. In this review, we integrate the results of previous studies and describe the cholesterol landscape in cellular homeostasis and neuronal function. We discuss its implication in α-Syn and Lewy body pathophysiological mechanisms underlying PD, focusing on the role of GCase and cholesterol. © 2020 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Keywords: autophagy; glycosphingolipid; lipid storage diseases; lysosomes; multilamellar bodies; neurodegeneration.
© 2020 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Figures
Similar articles
-
Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson's Patient Midbrain Neurons.J Neurosci. 2016 Jul 20;36(29):7693-706. doi: 10.1523/JNEUROSCI.0628-16.2016. J Neurosci. 2016. PMID: 27445146 Free PMC article.
-
Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease.Adv Drug Deliv Rev. 2022 Aug;187:114402. doi: 10.1016/j.addr.2022.114402. Epub 2022 Jun 25. Adv Drug Deliv Rev. 2022. PMID: 35764179 Review.
-
Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function.Hum Mol Genet. 2018 Jun 1;27(11):1972-1988. doi: 10.1093/hmg/ddy105. Hum Mol Genet. 2018. PMID: 29579237 Free PMC article.
-
A novel glucosylceramide synthase inhibitor attenuates alpha synuclein pathology and lysosomal dysfunction in preclinical models of synucleinopathy.Neurobiol Dis. 2021 Nov;159:105507. doi: 10.1016/j.nbd.2021.105507. Epub 2021 Sep 9. Neurobiol Dis. 2021. PMID: 34509608
-
GBA1 mutations: Prospects for exosomal biomarkers in α-synuclein pathologies.Mol Genet Metab. 2020 Feb;129(2):35-46. doi: 10.1016/j.ymgme.2019.10.006. Epub 2019 Oct 23. Mol Genet Metab. 2020. PMID: 31761523 Free PMC article. Review.
Cited by
-
Gut microbiota in combination with blood metabolites reveals characteristics of the disease cluster of coronary artery disease and cognitive impairment: a Mendelian randomization study.Front Immunol. 2024 Jan 15;14:1308002. doi: 10.3389/fimmu.2023.1308002. eCollection 2023. Front Immunol. 2024. PMID: 38288114 Free PMC article.
-
Metabolic Diffusion in Neuropathologies: The Relevance of Brain-Liver Axis.Front Physiol. 2022 May 12;13:864263. doi: 10.3389/fphys.2022.864263. eCollection 2022. Front Physiol. 2022. PMID: 35634148 Free PMC article. Review.
-
Neurodegenerative Diseases and Cholesterol: Seeing the Field Through the Players.Front Aging Neurosci. 2021 Nov 3;13:766587. doi: 10.3389/fnagi.2021.766587. eCollection 2021. Front Aging Neurosci. 2021. PMID: 34803658 Free PMC article. Review.
-
Lipid level alteration in human and cellular models of alpha synuclein mutations.NPJ Parkinsons Dis. 2022 Apr 25;8(1):52. doi: 10.1038/s41531-022-00313-y. NPJ Parkinsons Dis. 2022. PMID: 35468903 Free PMC article.
-
GCase Enhancers: A Potential Therapeutic Option for Gaucher Disease and Other Neurological Disorders.Pharmaceuticals (Basel). 2022 Jul 2;15(7):823. doi: 10.3390/ph15070823. Pharmaceuticals (Basel). 2022. PMID: 35890122 Free PMC article. Review.
References
-
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 2004;24:806–815. - PubMed
-
- Dietschy JM, Turley SD. Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 2004;45:1375–1397. - PubMed
-
- Friedrichson T, Kurzchalia TV. Microdomains of GPI‐anchored proteins in living cells revealed by crosslinking. Nature 1998;394:802–805. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
