

Mutations in Spliceosomal Genes PPIL1 and PRP17 Cause Neurodegenerative Pontocerebellar Hypoplasia with Microcephaly
- PMID: 33220177
- PMCID: PMC8800389
- DOI: 10.1016/j.neuron.2020.10.035
Mutations in Spliceosomal Genes PPIL1 and PRP17 Cause Neurodegenerative Pontocerebellar Hypoplasia with Microcephaly
Abstract
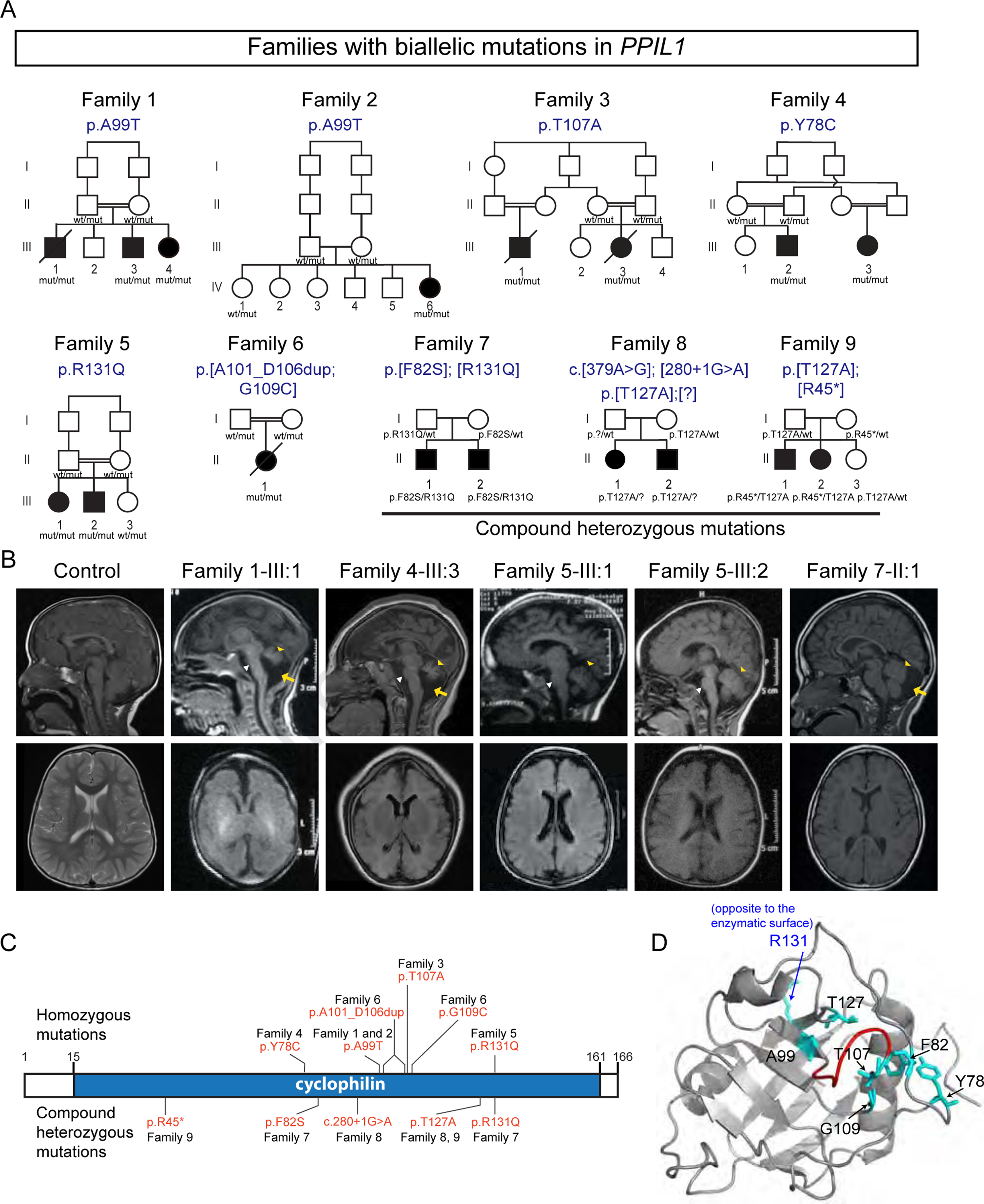
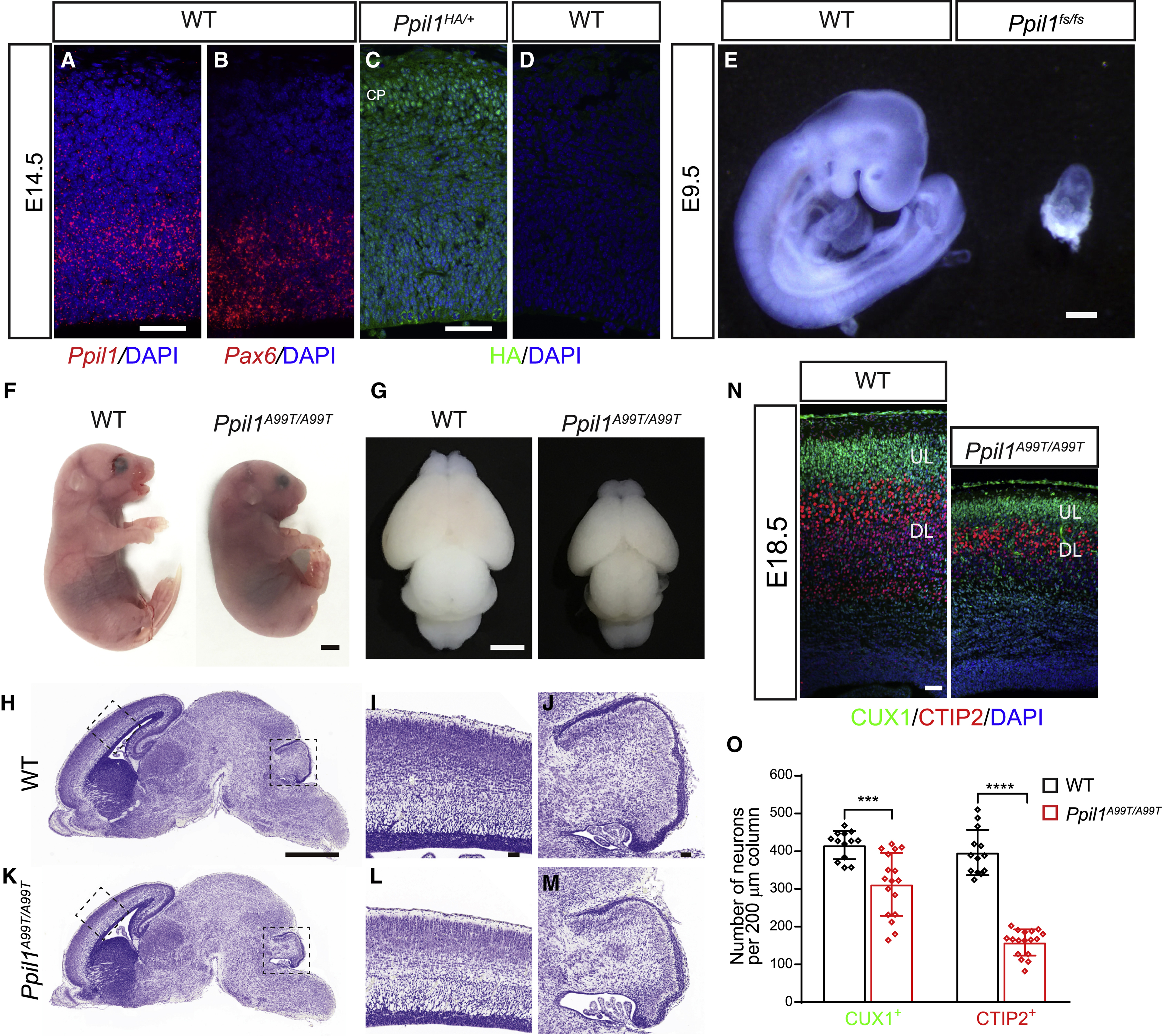
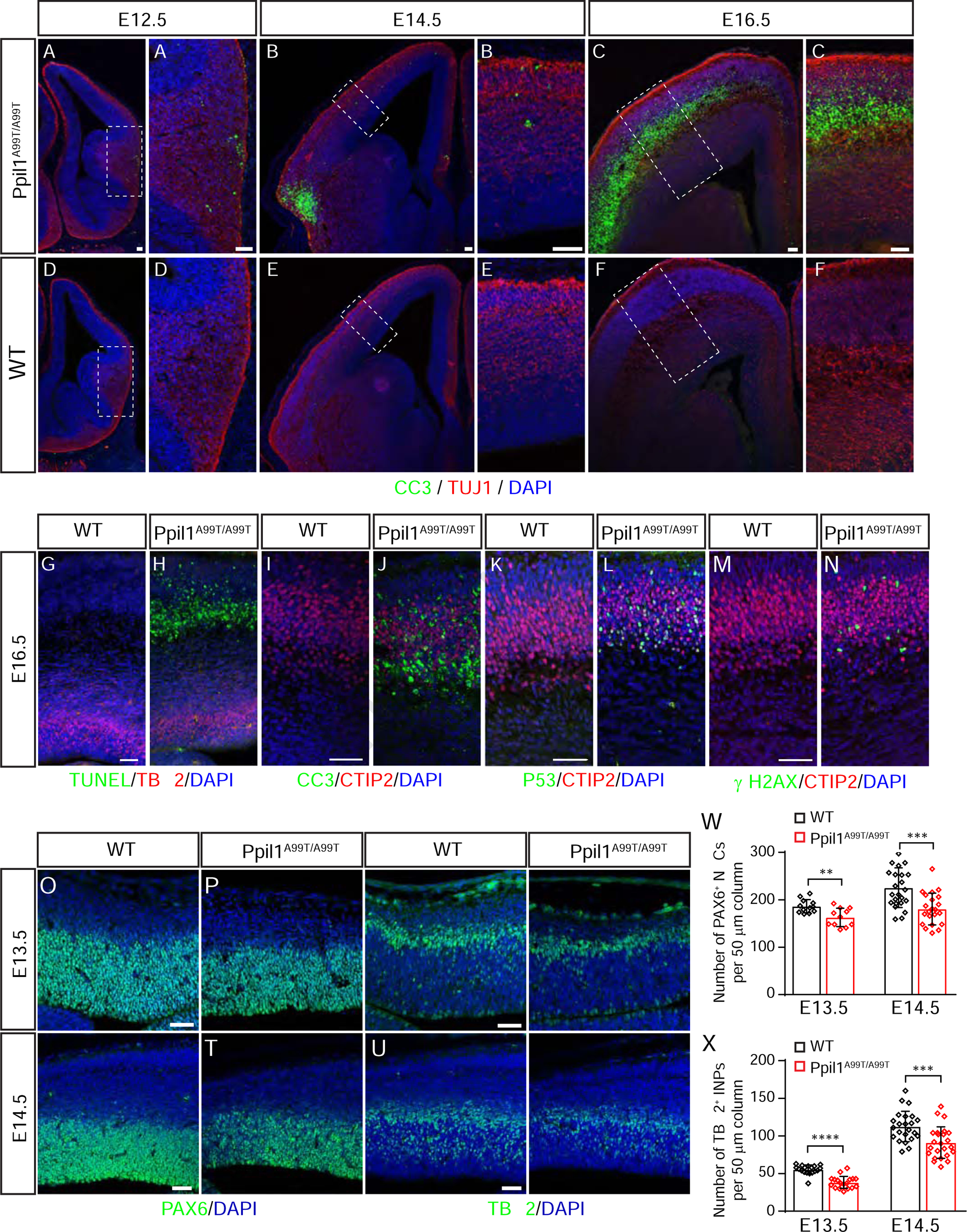
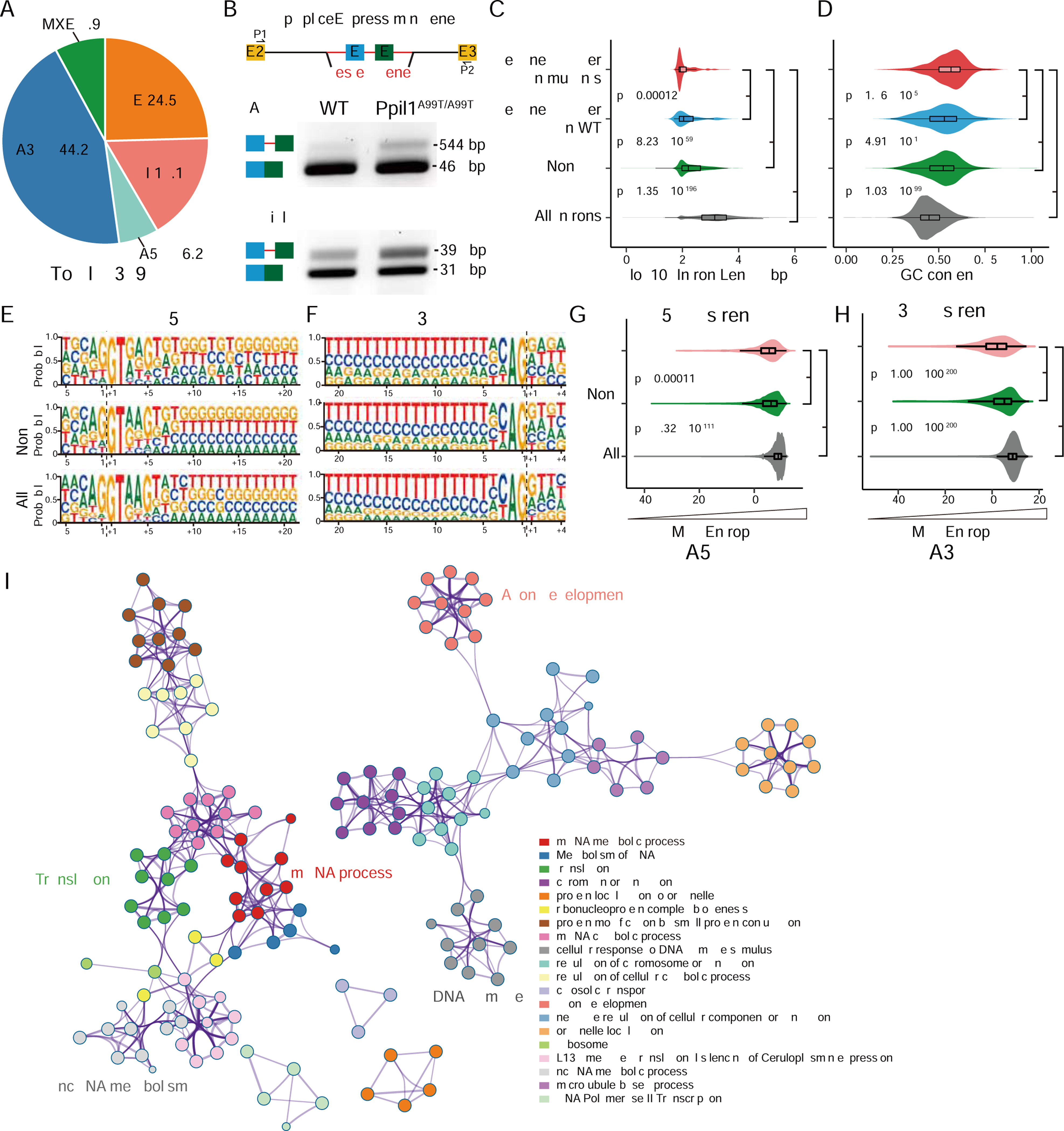
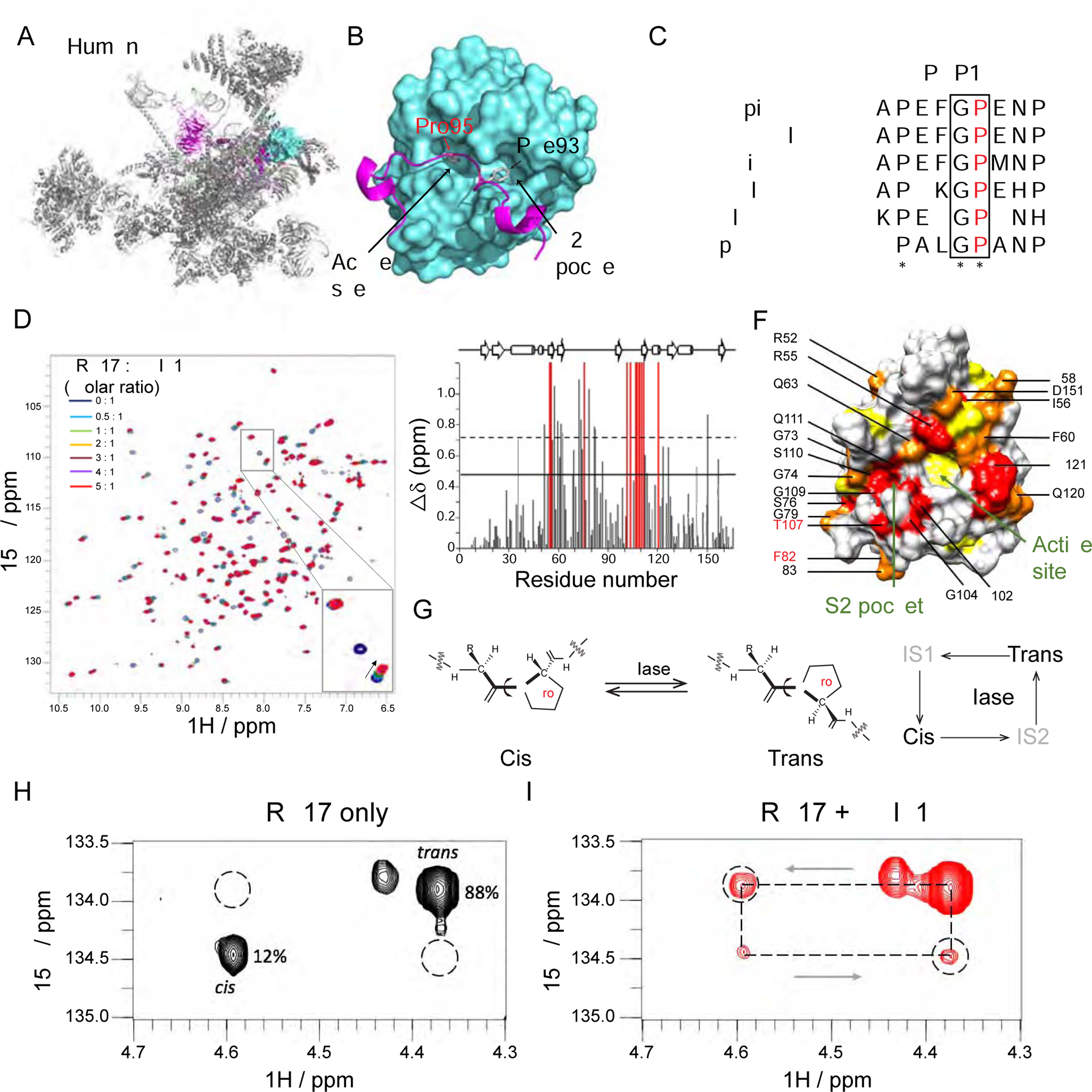
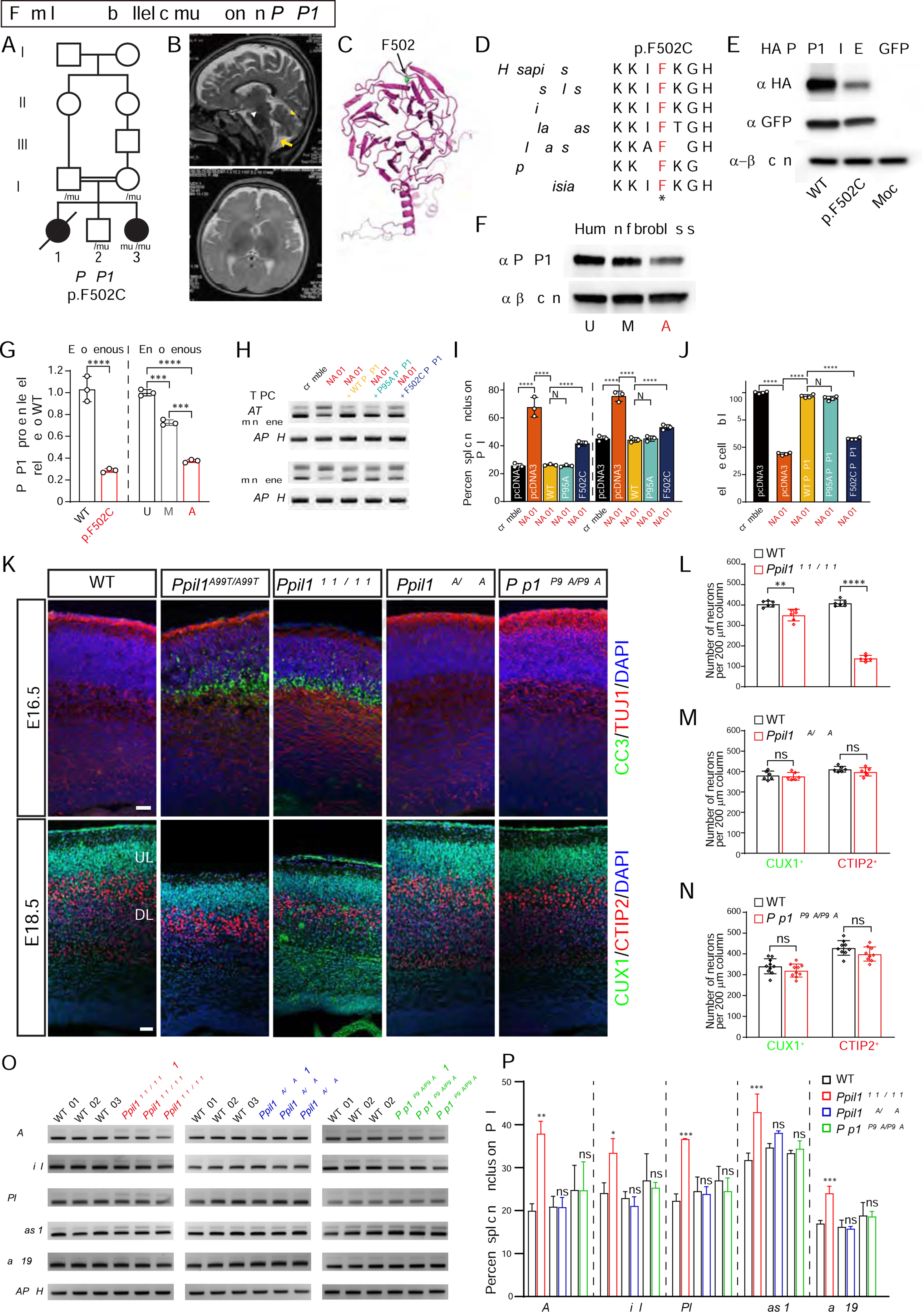
Autosomal-recessive cerebellar hypoplasia and ataxia constitute a group of heterogeneous brain disorders caused by disruption of several fundamental cellular processes. Here, we identified 10 families showing a neurodegenerative condition involving pontocerebellar hypoplasia with microcephaly (PCHM). Patients harbored biallelic mutations in genes encoding the spliceosome components Peptidyl-Prolyl Isomerase Like-1 (PPIL1) or Pre-RNA Processing-17 (PRP17). Mouse knockouts of either gene were lethal in early embryogenesis, whereas PPIL1 patient mutation knockin mice showed neuron-specific apoptosis. Loss of either protein affected splicing integrity, predominantly affecting short and high GC-content introns and genes involved in brain disorders. PPIL1 and PRP17 form an active isomerase-substrate interaction, but we found that isomerase activity is not critical for function. Thus, we establish disrupted splicing integrity and "major spliceosome-opathies" as a new mechanism underlying PCHM and neurodegeneration and uncover a non-enzymatic function of a spliceosomal proline isomerase.
Keywords: NMR; PCHM; PPIL1; PRP17; alternative splicing; brain development; cyclophilin; microcephaly; neurodegeneration; pontocerebellar hypoplasia; proline isomerase; recessive disease; spliceosome.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Comment in
-
Splicing Control of Pontocerebellar Development.Neuron. 2021 Jan 20;109(2):191-192. doi: 10.1016/j.neuron.2020.12.021. Neuron. 2021. PMID: 33476558
References
-
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, and Luhrmann R. (2017). Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542, 318–323. - PubMed
-
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, and Luhrmann R. (2010). Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16, 2384–2403. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- R01 NS092772/NS/NINDS NIH HHS/United States
- F32 NS009800/NS/NINDS NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- UL1 TR001442/TR/NCATS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 NS048453/NS/NINDS NIH HHS/United States
- 094232/WT_/Wellcome Trust/United Kingdom
- U54 HG006504/HG/NHGRI NIH HHS/United States
- S10 OD026929/OD/NIH HHS/United States
- MR/K011154/1/MRC_/Medical Research Council/United Kingdom
- R01 NS098004/NS/NINDS NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- MR/M009084/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
