

Microbial function and genital inflammation in young South African women at high risk of HIV infection
- PMID: 33220709
- PMCID: PMC7679981
- DOI: 10.1186/s40168-020-00932-8
Microbial function and genital inflammation in young South African women at high risk of HIV infection
Erratum in
-
Correction to: Microbial function and genital inflammation in young South African women at high risk of HIV infection.Microbiome. 2022 Mar 9;10(1):42. doi: 10.1186/s40168-022-01245-8. Microbiome. 2022. PMID: 35264249 Free PMC article. No abstract available.
Abstract
Background: Female genital tract (FGT) inflammation is an important risk factor for HIV acquisition. The FGT microbiome is closely associated with inflammatory profile; however, the relative importance of microbial activities has not been established. Since proteins are key elements representing actual microbial functions, this study utilized metaproteomics to evaluate the relationship between FGT microbial function and inflammation in 113 young and adolescent South African women at high risk of HIV infection. Women were grouped as having low, medium, or high FGT inflammation by K-means clustering according to pro-inflammatory cytokine concentrations.
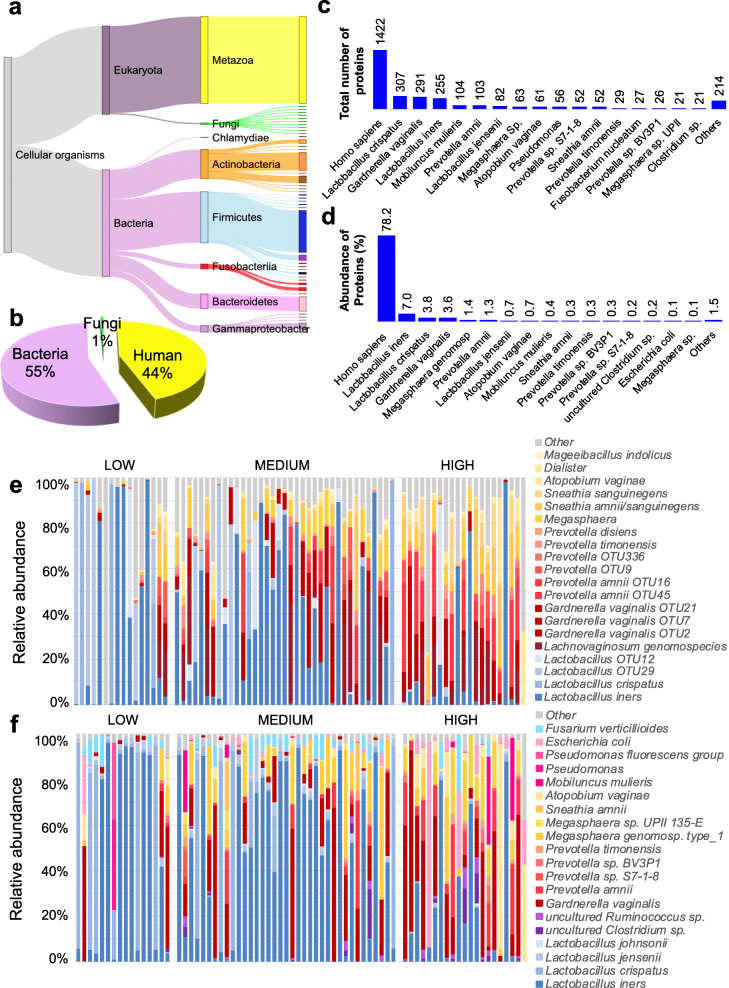
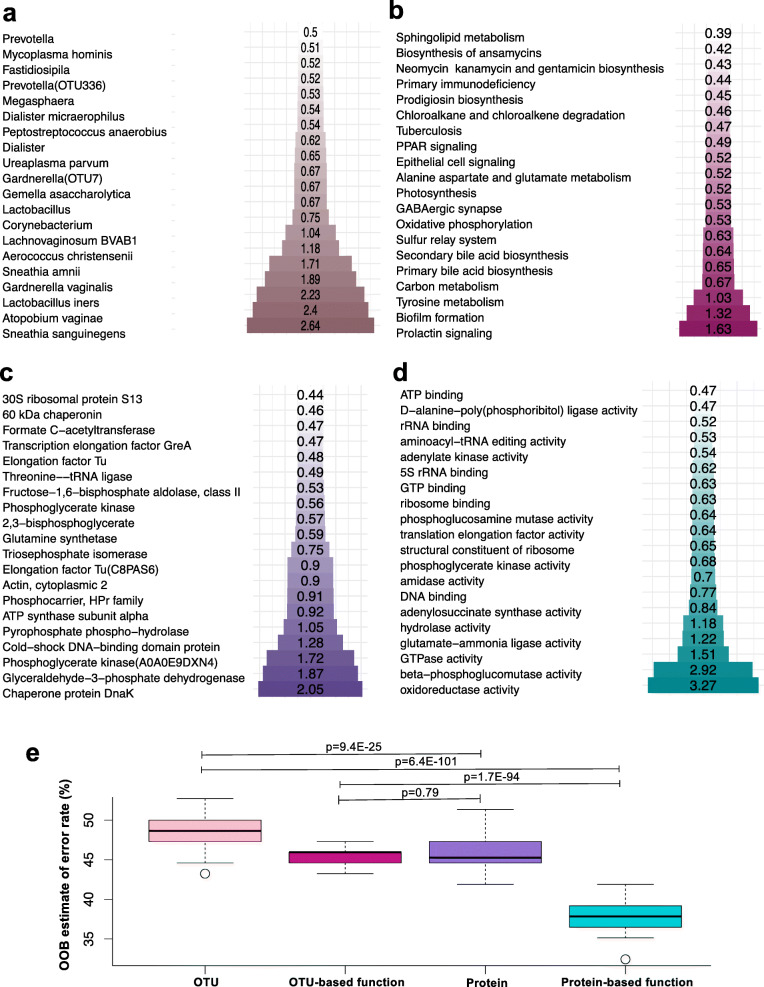
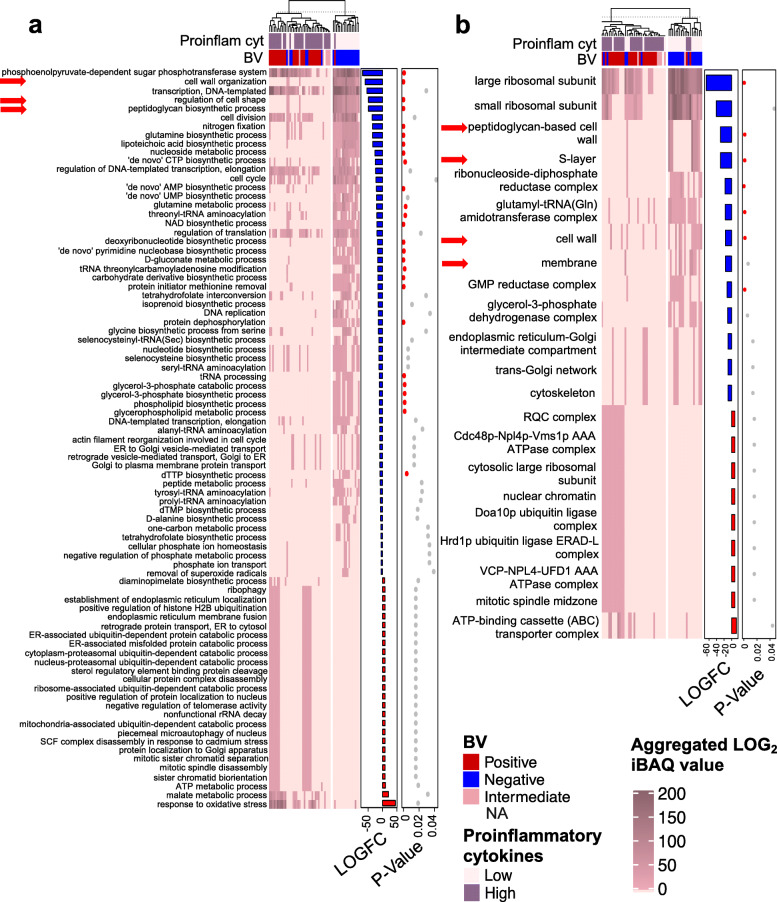
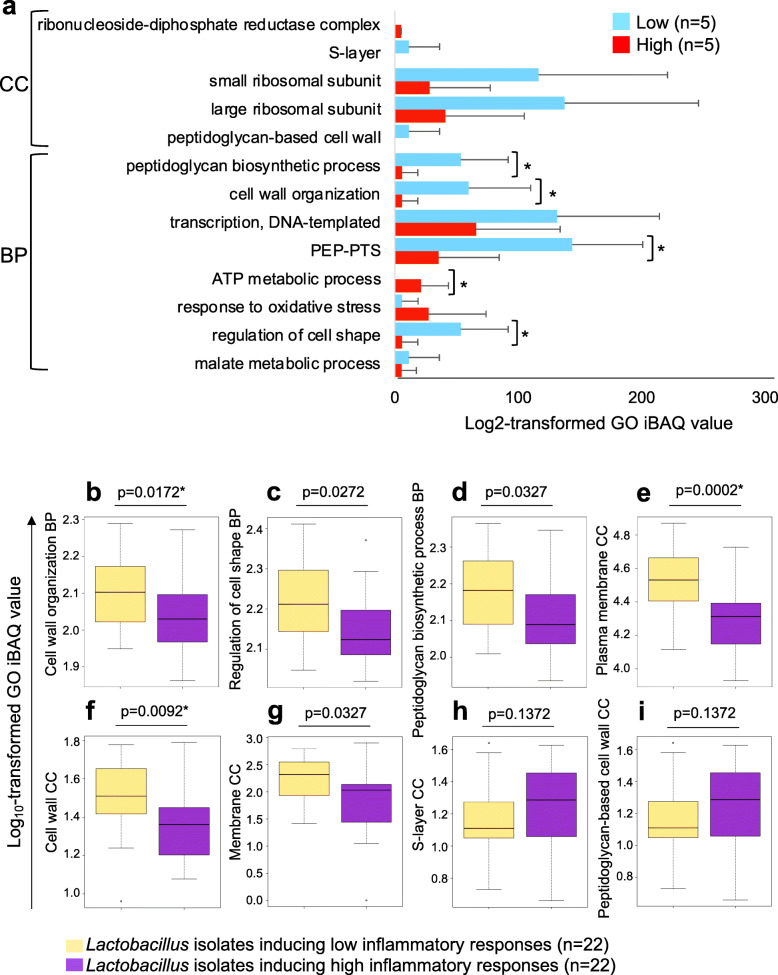
Results: A total of 3186 microbial and human proteins were identified in lateral vaginal wall swabs using liquid chromatography-tandem mass spectrometry, while 94 microbial taxa were included in the taxonomic analysis. Both metaproteomics and 16S rRNA gene sequencing analyses showed increased non-optimal bacteria and decreased lactobacilli in women with FGT inflammatory profiles. However, differences in the predicted relative abundance of most bacteria were observed between 16S rRNA gene sequencing and metaproteomics analyses. Bacterial protein functional annotations (gene ontology) predicted inflammatory cytokine profiles more accurately than bacterial relative abundance determined by 16S rRNA gene sequence analysis, as well as functional predictions based on 16S rRNA gene sequence data (p < 0.0001). The majority of microbial biological processes were underrepresented in women with high inflammation compared to those with low inflammation, including a Lactobacillus-associated signature of reduced cell wall organization and peptidoglycan biosynthesis. This signature remained associated with high FGT inflammation in a subset of 74 women 9 weeks later, was upheld after adjusting for Lactobacillus relative abundance, and was associated with in vitro inflammatory cytokine responses to Lactobacillus isolates from the same women. Reduced cell wall organization and peptidoglycan biosynthesis were also associated with high FGT inflammation in an independent sample of ten women.
Conclusions: Both the presence of specific microbial taxa in the FGT and their properties and activities are critical determinants of FGT inflammation. Our findings support those of previous studies suggesting that peptidoglycan is directly immunosuppressive, and identify a possible avenue for biotherapeutic development to reduce inflammation in the FGT. To facilitate further investigations of microbial activities, we have developed the FGT-DB application that is available at http://fgtdb.org/ . Video Abstract.
Keywords: Cytokine; Female genital tract; Inflammation; Metaproteomics; Microbial function; Microbiome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- UNAIDS Data 2019. Available at: https://www.unaids.org/sites/default/files/media_asset/2019-UNAIDS-data_.... Accessed 20/02/20.
-
- Morrison C, Fichorova RN, Mauck C, Chen PL, Kwok C, Chipato T, et al. Cervical inflammation and immunity associated with hormonal contraception, pregnancy, and HIV-1 seroconversion. J Acquir Immune Defic Syndr. 2014;66:109–117. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
