

Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review
- PMID: 33224105
- PMCID: PMC7674493
- DOI: 10.3389/fendo.2020.560375
Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review
Abstract
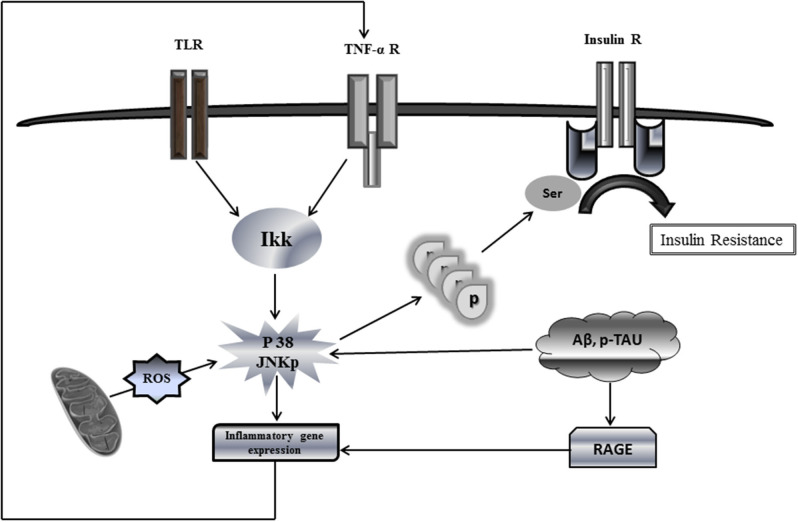
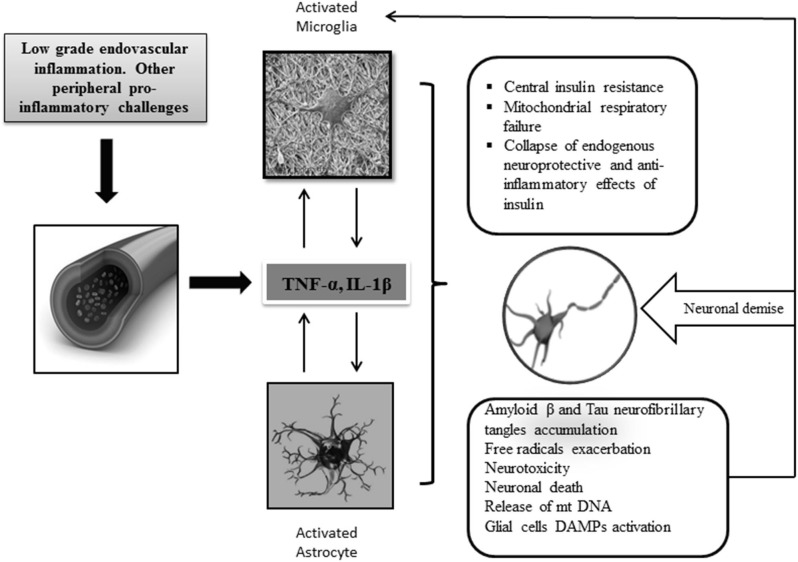
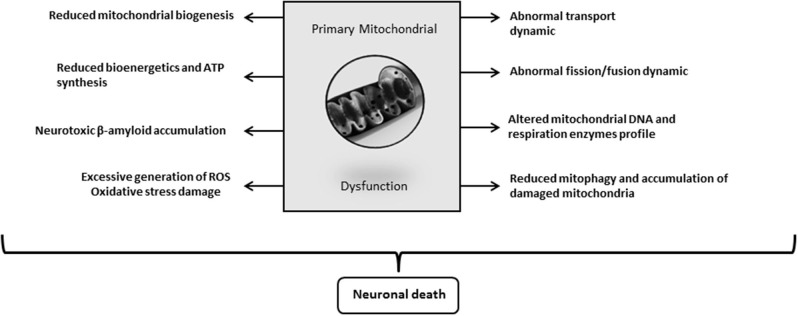
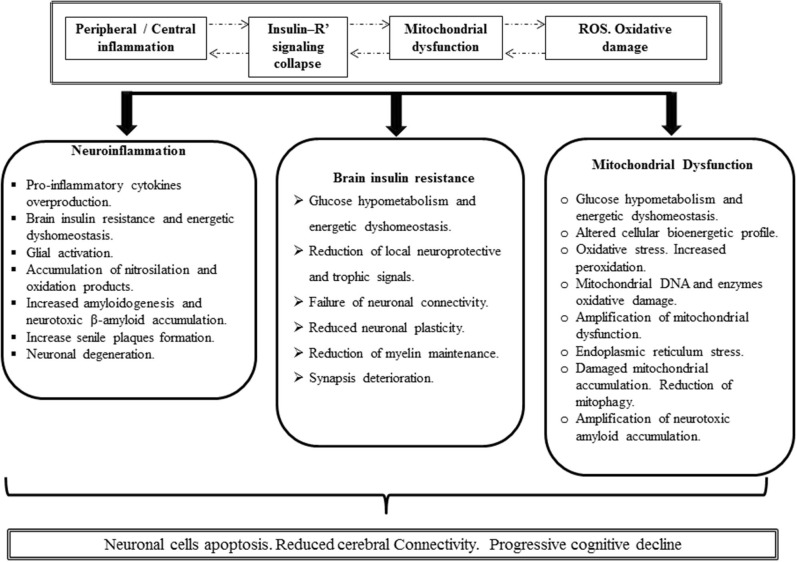
Insulin plays a major neuroprotective and trophic function for cerebral cell population, thus countering apoptosis, beta-amyloid toxicity, and oxidative stress; favoring neuronal survival; and enhancing memory and learning processes. Insulin resistance and impaired cerebral glucose metabolism are invariantly reported in Alzheimer's disease (AD) and other neurodegenerative processes. AD is a fatal neurodegenerative disorder in which progressive glucose hypometabolism parallels to cognitive impairment. Although AD may appear and progress in virtue of multifactorial nosogenic ingredients, multiple interperpetuative and interconnected vicious circles appear to drive disease pathophysiology. The disease is primarily a metabolic/energetic disorder in which amyloid accumulation may appear as a by-product of more proximal events, especially in the late-onset form. As a bridge between AD and type 2 diabetes, activation of c-Jun N-terminal kinase (JNK) pathway with the ensued serine phosphorylation of the insulin response substrate (IRS)-1/2 may be at the crossroads of insulin resistance and its subsequent dysmetabolic consequences. Central insulin axis bankruptcy translates in neuronal vulnerability and demise. As a link in the chain of pathogenic vicious circles, mitochondrial dysfunction, oxidative stress, and peripheral/central immune-inflammation are increasingly advocated as major pathology drivers. Pharmacological interventions addressed to preserve insulin axis physiology, mitochondrial biogenesis-integral functionality, and mitophagy of diseased organelles may attenuate the adjacent spillover of free radicals that further perpetuate mitochondrial damages and catalyze inflammation. Central and/or peripheral inflammation may account for a local flood of proinflammatory cytokines that along with astrogliosis amplify insulin resistance, mitochondrial dysfunction, and oxidative stress. All these elements are endogenous stressor, pro-senescent factors that contribute to JNK activation. Taken together, these evidences incite to identify novel multi-mechanistic approaches to succeed in ameliorating this pandemic affliction.
Keywords: Alzheimer's disease; central insuline resistance; neurodegeneration; neurofibrillary tangles; β-amyloid plaques.
Copyright © 2020 Berlanga-Acosta, Guillén-Nieto, Rodríguez-Rodríguez, Bringas-Vega, García-del-Barco-Herrera, Berlanga-Saez, García-Ojalvo, Valdés-Sosa and Valdés-Sosa.
Figures
Similar articles
-
Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies.Prog Neurobiol. 2013 Sep;108:21-43. doi: 10.1016/j.pneurobio.2013.06.004. Epub 2013 Jul 11. Prog Neurobiol. 2013. PMID: 23850509 Review.
-
Insulin signaling: An opportunistic target to minify the risk of Alzheimer's disease.Psychoneuroendocrinology. 2017 Sep;83:159-171. doi: 10.1016/j.psyneuen.2017.05.004. Epub 2017 May 30. Psychoneuroendocrinology. 2017. PMID: 28624654 Review.
-
Role of insulin resistance in Alzheimer's disease.Metab Brain Dis. 2015 Aug;30(4):839-51. doi: 10.1007/s11011-014-9631-3. Epub 2014 Nov 16. Metab Brain Dis. 2015. PMID: 25399337 Review.
-
Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer's disease.Biochim Biophys Acta. 2009 May;1792(5):482-96. doi: 10.1016/j.bbadis.2008.10.014. Epub 2008 Nov 5. Biochim Biophys Acta. 2009. PMID: 19026743 Review.
-
Streptozotocin Intracerebroventricular-Induced Neurotoxicity and Brain Insulin Resistance: a Therapeutic Intervention for Treatment of Sporadic Alzheimer's Disease (sAD)-Like Pathology.Mol Neurobiol. 2016 Sep;53(7):4548-62. doi: 10.1007/s12035-015-9384-y. Epub 2015 Aug 23. Mol Neurobiol. 2016. PMID: 26298663 Review.
Cited by
-
Therapeutic Efficacy of the Inositol D-Pinitol as a Multi-Faceted Disease Modifier in the 5×FAD Humanized Mouse Model of Alzheimer's Amyloidosis.Nutrients. 2024 Dec 4;16(23):4186. doi: 10.3390/nu16234186. Nutrients. 2024. PMID: 39683582 Free PMC article.
-
Multi-Ancestry Transcriptome-Wide Association Studies of Cognitive Function, White Matter Hyperintensity, and Alzheimer's Disease.Int J Mol Sci. 2025 Mar 9;26(6):2443. doi: 10.3390/ijms26062443. Int J Mol Sci. 2025. PMID: 40141087 Free PMC article.
-
Pharmacological Approaches to the Treatment of Dementia in Down Syndrome: A Systematic Review of Randomized Clinical Studies.Molecules. 2022 May 19;27(10):3244. doi: 10.3390/molecules27103244. Molecules. 2022. PMID: 35630721 Free PMC article.
-
Cyclosorus Terminans Extract Alleviates Neuroinflammation in Insulin Resistant Rats.Mol Neurobiol. 2024 Jul;61(7):4879-4890. doi: 10.1007/s12035-023-03883-x. Epub 2023 Dec 27. Mol Neurobiol. 2024. PMID: 38148371
-
Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer's Disease.Int J Mol Sci. 2021 May 25;22(11):5603. doi: 10.3390/ijms22115603. Int J Mol Sci. 2021. PMID: 34070553 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous