

New developments in Huntington's disease and other triplet repeat diseases: DNA repair turns to the dark side
- PMID: 33224521
- PMCID: PMC7672267
- DOI: 10.1042/NS20200010
New developments in Huntington's disease and other triplet repeat diseases: DNA repair turns to the dark side
Abstract
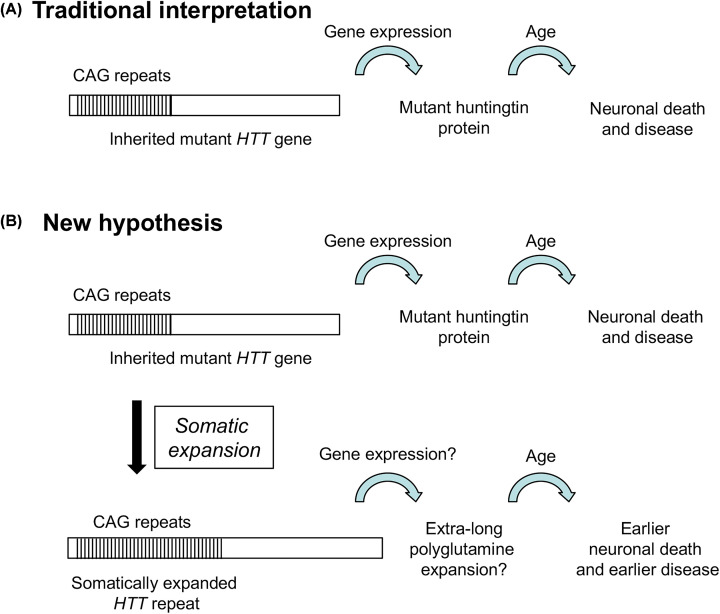
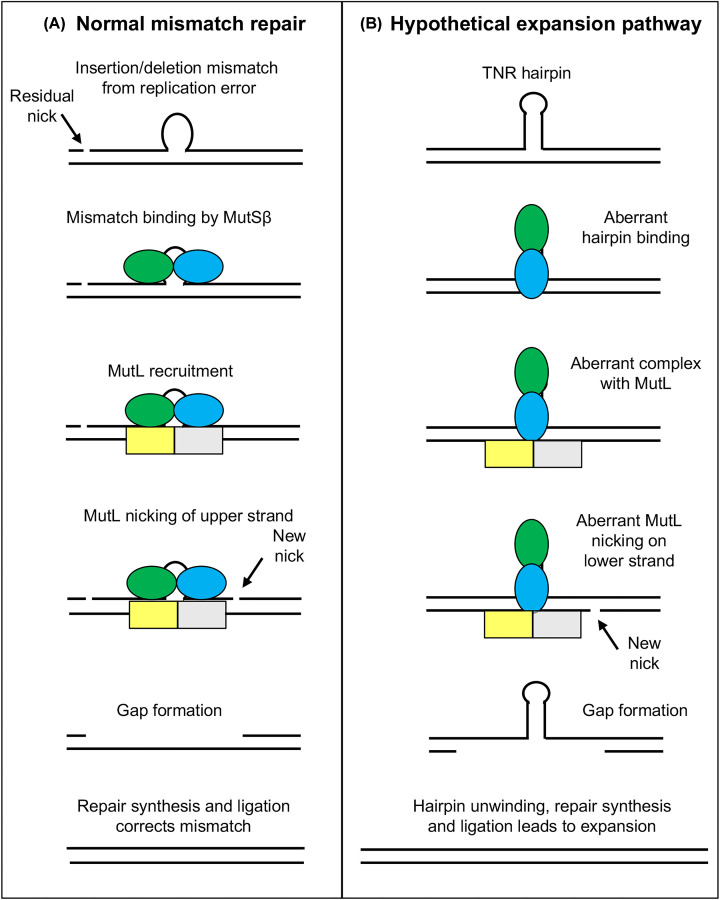
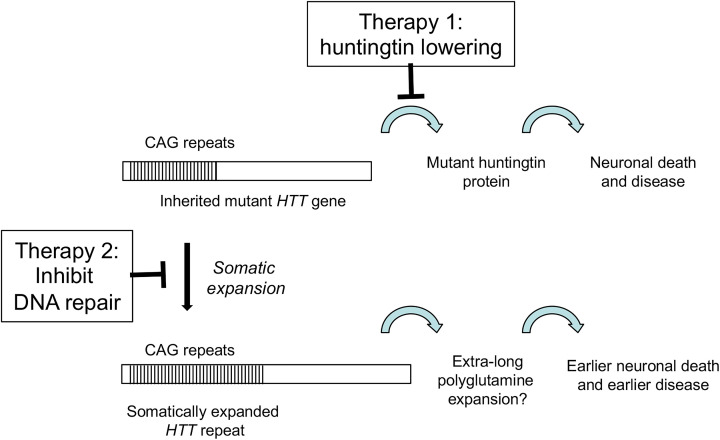
Huntington's disease (HD) is a fatal, inherited neurodegenerative disease that causes neuronal death, particularly in medium spiny neurons. HD leads to serious and progressive motor, cognitive and psychiatric symptoms. Its genetic basis is an expansion of the CAG triplet repeat in the HTT gene, leading to extra glutamines in the huntingtin protein. HD is one of nine genetic diseases in this polyglutamine (polyQ) category, that also includes a number of inherited spinocerebellar ataxias (SCAs). Traditionally it has been assumed that HD age of onset and disease progression were solely the outcome of age-dependent exposure of neurons to toxic effects of the inherited mutant huntingtin protein. However, recent genome-wide association studies (GWAS) have revealed significant effects of genetic variants outside of HTT. Surprisingly, these variants turn out to be mostly in genes encoding DNA repair factors, suggesting that at least some disease modulation occurs at the level of the HTT DNA itself. These DNA repair proteins are known from model systems to promote ongoing somatic CAG repeat expansions in tissues affected by HD. Thus, for triplet repeats, some DNA repair proteins seem to abandon their normal genoprotective roles and, instead, drive expansions and accelerate disease. One attractive hypothesis-still to be proven rigorously-is that somatic HTT expansions augment the disease burden of the inherited allele. If so, therapeutic approaches that lower levels of huntingtin protein may need blending with additional therapies that reduce levels of somatic CAG repeat expansions to achieve maximal effect.
Keywords: DNA synthesis and repair; genome integrity; mutation; neurodegeneration.
© 2020 The Author(s).
Conflict of interest statement
The author is a paid consultant of LoQus23 Therapeutics and serves on their scientific advisory board; and Science Foundation Ireland [grant number 16/BBSRC/3395].The authors declare that there are no competing interests associated with the manuscript.
Figures

Similar articles
-
Multiple clinical features of Huntington's disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration.J Neurol. 2019 Mar;266(3):551-564. doi: 10.1007/s00415-018-8940-6. Epub 2018 Jun 28. J Neurol. 2019. PMID: 29956026 Review.
-
PMS1 as a target for splice modulation to prevent somatic CAG repeat expansion in Huntington's disease.bioRxiv [Preprint]. 2023 Jul 27:2023.07.25.550489. doi: 10.1101/2023.07.25.550489. bioRxiv. 2023. Update in: Nat Commun. 2024 Apr 12;15(1):3182. doi: 10.1038/s41467-024-47485-0. PMID: 37547003 Free PMC article. Updated. Preprint.
-
Interrupting sequence variants and age of onset in Huntington's disease: clinical implications and emerging therapies.Lancet Neurol. 2020 Nov;19(11):930-939. doi: 10.1016/S1474-4422(20)30343-4. Lancet Neurol. 2020. PMID: 33098802 Review.
-
Huntington’s Disease Pathogenesis: Mechanisms and Pathways.In: Lo DC, Hughes RE, editors. Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton (FL): CRC Press/Taylor & Francis; 2011. Chapter 2. In: Lo DC, Hughes RE, editors. Neurobiology of Huntington's Disease: Applications to Drug Discovery. Boca Raton (FL): CRC Press/Taylor & Francis; 2011. Chapter 2. PMID: 21882412 Free Books & Documents. Review.
-
Modification of Huntington's disease by short tandem repeats.Brain Commun. 2024 Jan 23;6(2):fcae016. doi: 10.1093/braincomms/fcae016. eCollection 2024. Brain Commun. 2024. PMID: 38449714 Free PMC article.
Cited by
-
Neuropathogenesis-on-chips for neurodegenerative diseases.Nat Commun. 2024 Mar 12;15(1):2219. doi: 10.1038/s41467-024-46554-8. Nat Commun. 2024. PMID: 38472255 Free PMC article. Review.
-
Msh2-Msh3 DNA-binding is not sufficient to promote trinucleotide repeat expansions in Saccharomyces cerevisiae.Genetics. 2025 Mar 17;229(3):iyae222. doi: 10.1093/genetics/iyae222. Genetics. 2025. PMID: 39790027 Free PMC article.
-
Neurodevelopmental Clues to Neurodegeneration.Pediatr Neurol. 2021 Oct;123:67-76. doi: 10.1016/j.pediatrneurol.2021.07.012. Epub 2021 Aug 6. Pediatr Neurol. 2021. PMID: 34399111 Free PMC article.
-
Neuronal Signaling: A reflection on the Biochemical Society's newest journal and an exciting outlook on its next steps.Neuronal Signal. 2021 Feb 8;5(1):NS20210007. doi: 10.1042/NS20210007. eCollection 2021 Apr. Neuronal Signal. 2021. PMID: 33585040 Free PMC article.
-
The Startling Role of Mismatch Repair in Trinucleotide Repeat Expansions.Cells. 2021 Apr 26;10(5):1019. doi: 10.3390/cells10051019. Cells. 2021. PMID: 33925919 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources