

Portopulmonary Hypertension: From Bench to Bedside
- PMID: 33224960
- PMCID: PMC7670077
- DOI: 10.3389/fmed.2020.569413
Portopulmonary Hypertension: From Bench to Bedside
Abstract
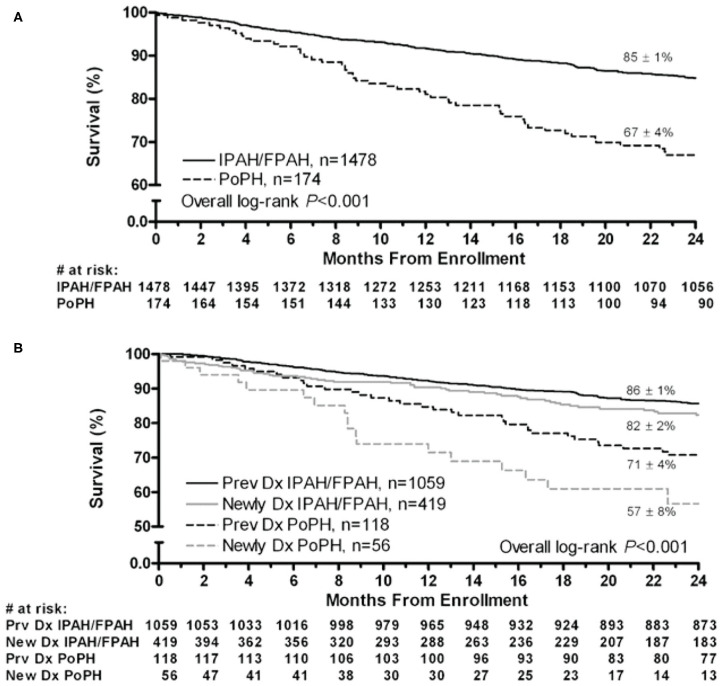
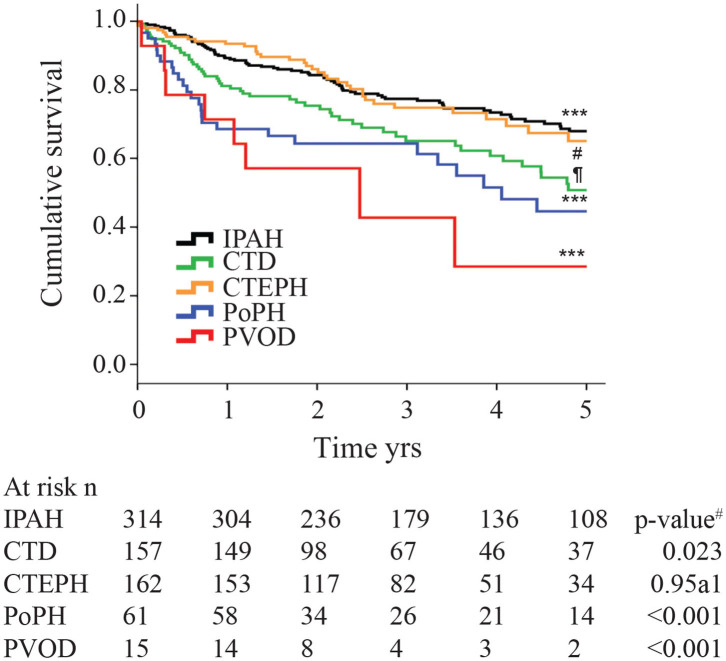
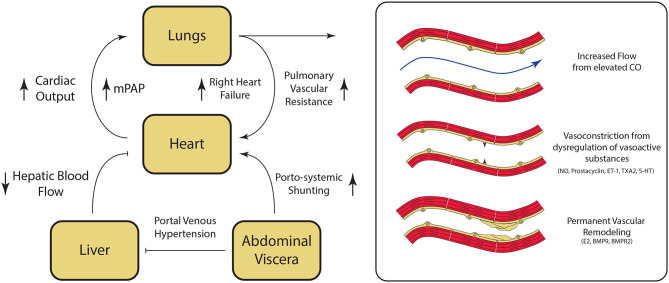
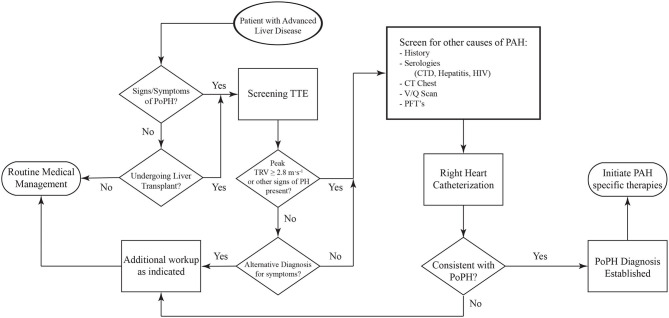
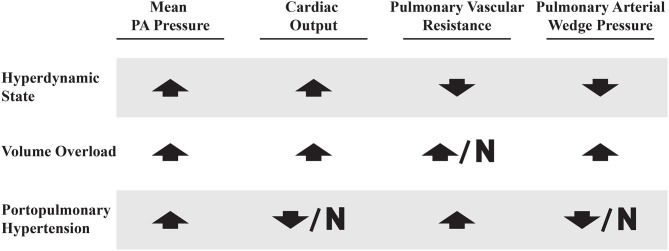
Portopulmonary hypertension (PoPH) is defined as pulmonary arterial hypertension (PAH) associated with portal hypertension and is a subset of Group 1 pulmonary hypertension (PH). PoPH is a cause of significant morbidity and mortality in patients with portal hypertension with or without liver disease. Significant strides in elucidating the pathogenesis, effective screening algorithms, accurate diagnoses, and treatment options have been made in past 20 years. Survival of PoPH has remained poor compared to IPAH and other forms of PAH. Recently, the first randomized controlled trial was done in this patient population and showed promising results with PAH specific therapy. Despite positive effects on hemodynamics and functional outcomes, it is unclear whether PAH specific therapy has a beneficial effect on long term survival or transplant outcomes. In this review, we will discuss the epidemiology, pathophysiology, clinical and hemodynamic characteristics of PoPH. Additionally, this review will highlight the lacunae in our current management strategy, challenges faced and will provide direction to potentially useful futuristic management strategies.
Keywords: MELD exception; liver transplant; portal hypertension; portopulmonary hypertension; pulmonary arterial hypertension.
Copyright © 2020 Thomas, Glinskii, de Jesus Perez and Sahay.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous