

Combined transient ablation and single-cell RNA-sequencing reveals the development of medullary thymic epithelial cells
- PMID: 33226342
- PMCID: PMC7771965
- DOI: 10.7554/eLife.60188
Combined transient ablation and single-cell RNA-sequencing reveals the development of medullary thymic epithelial cells
Abstract
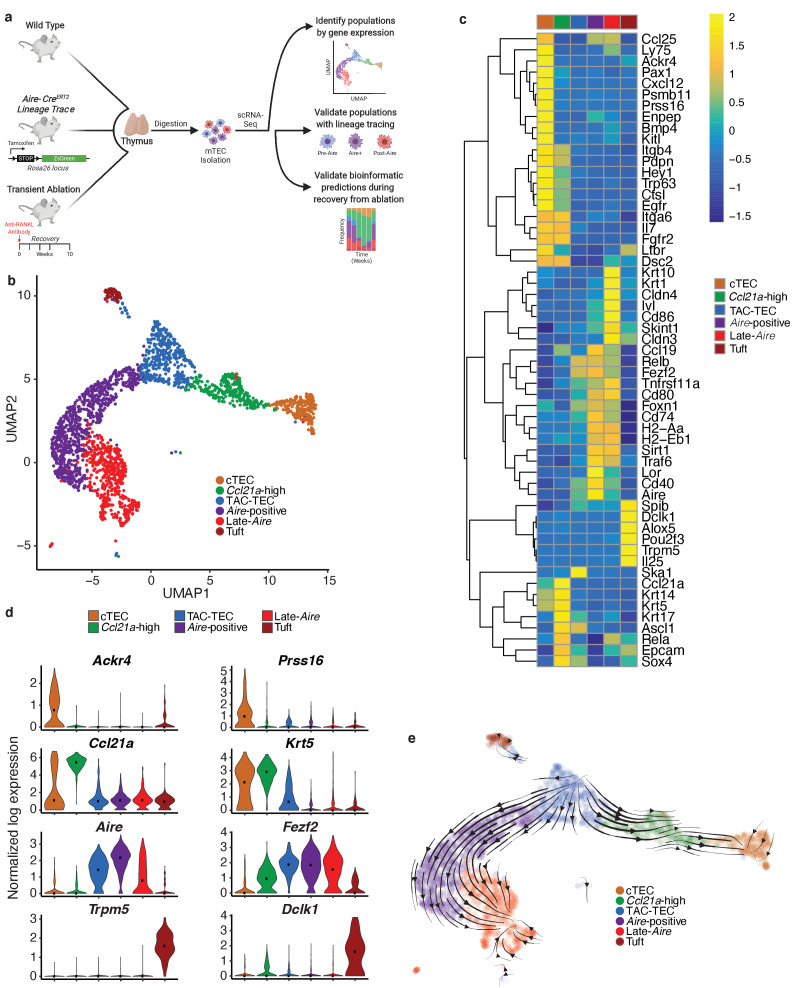
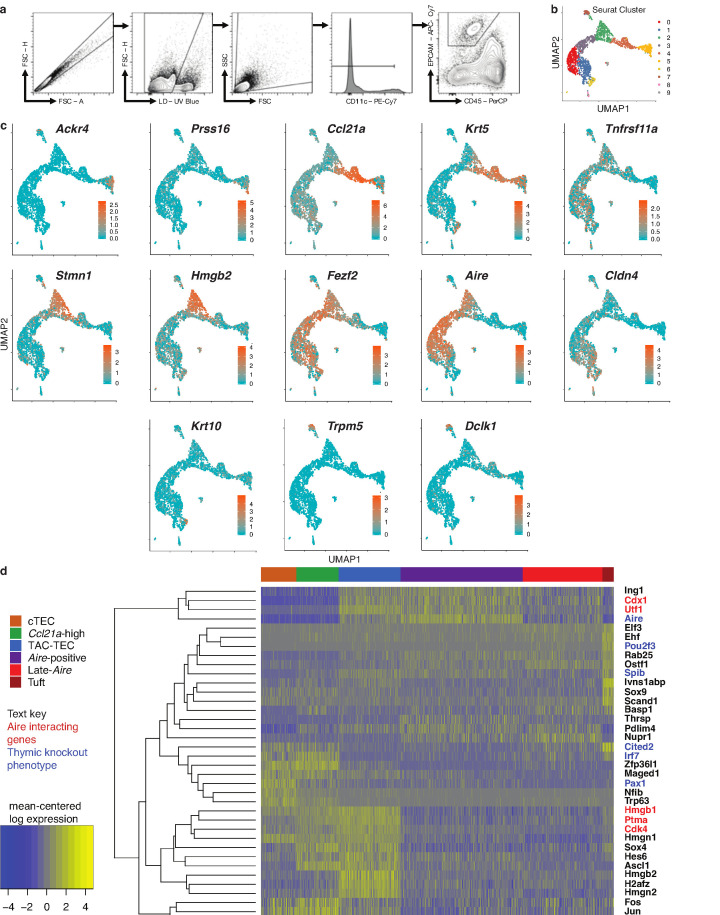
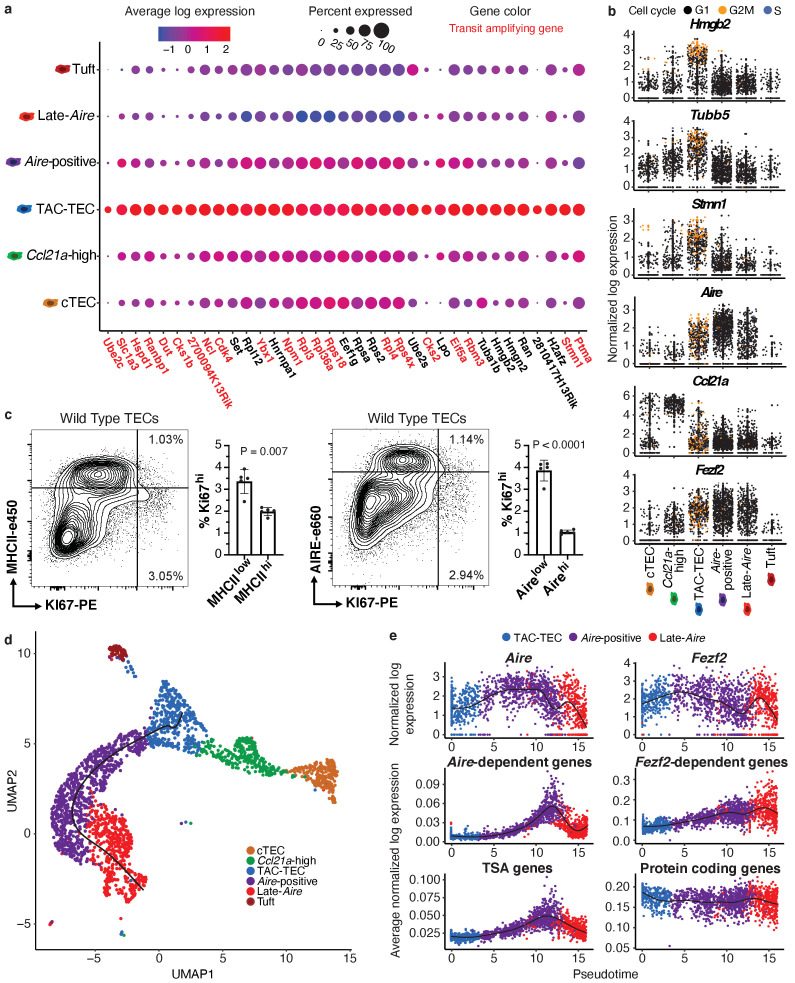
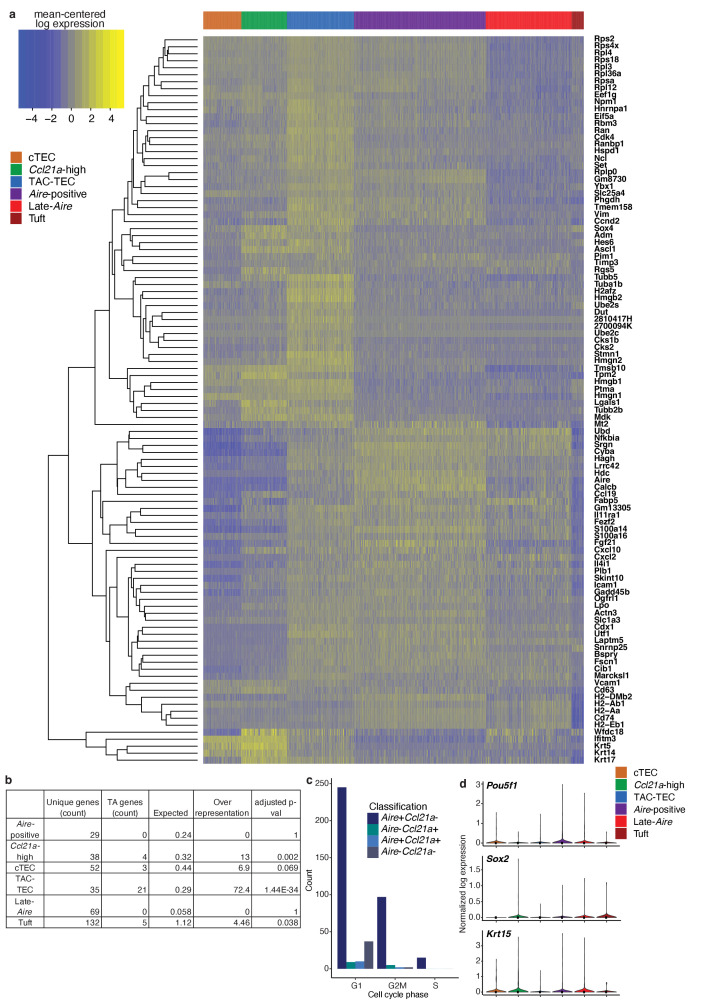
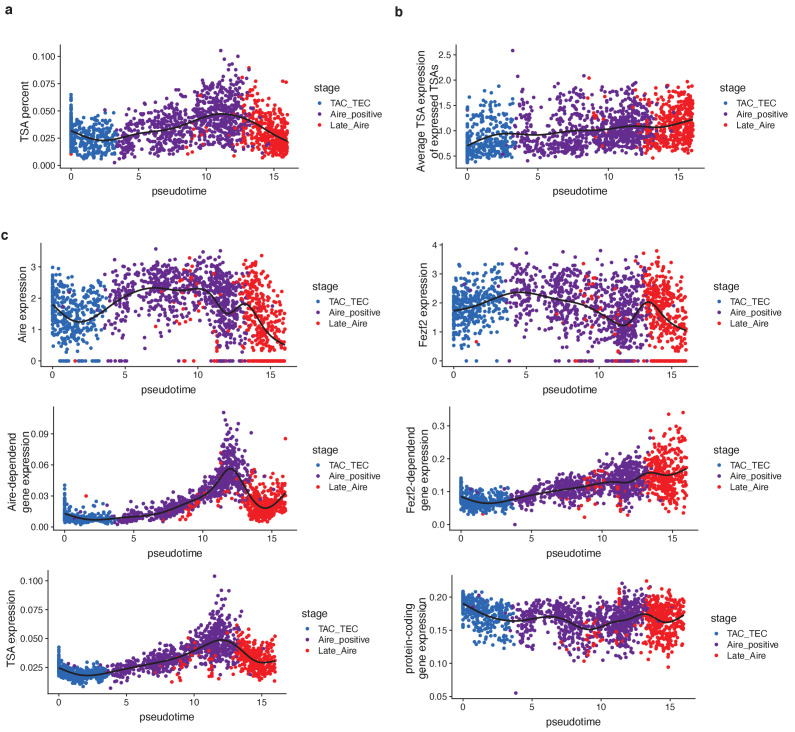
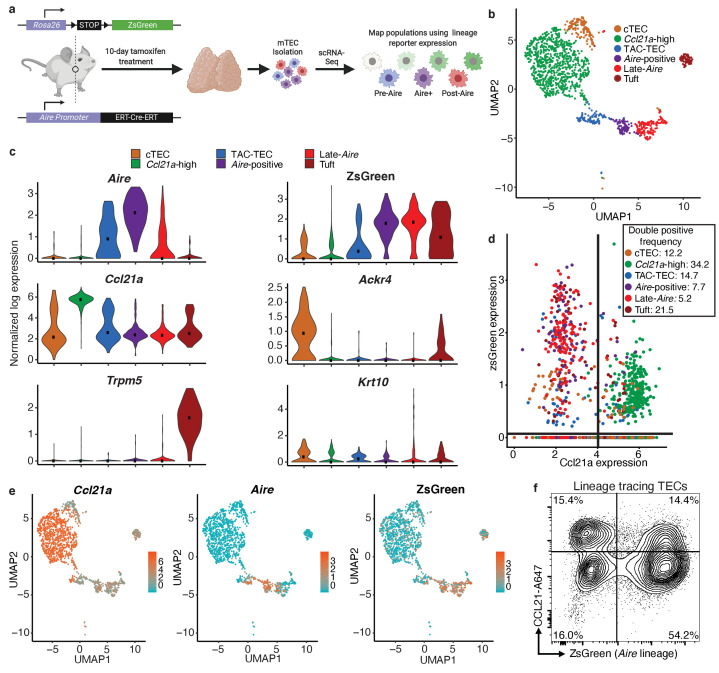
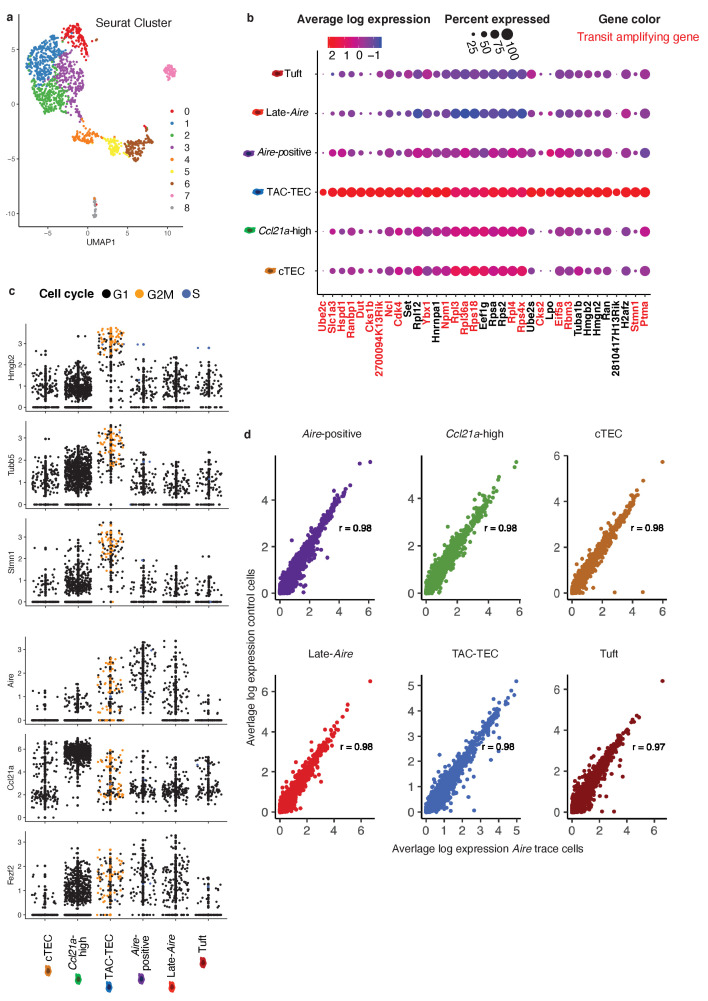
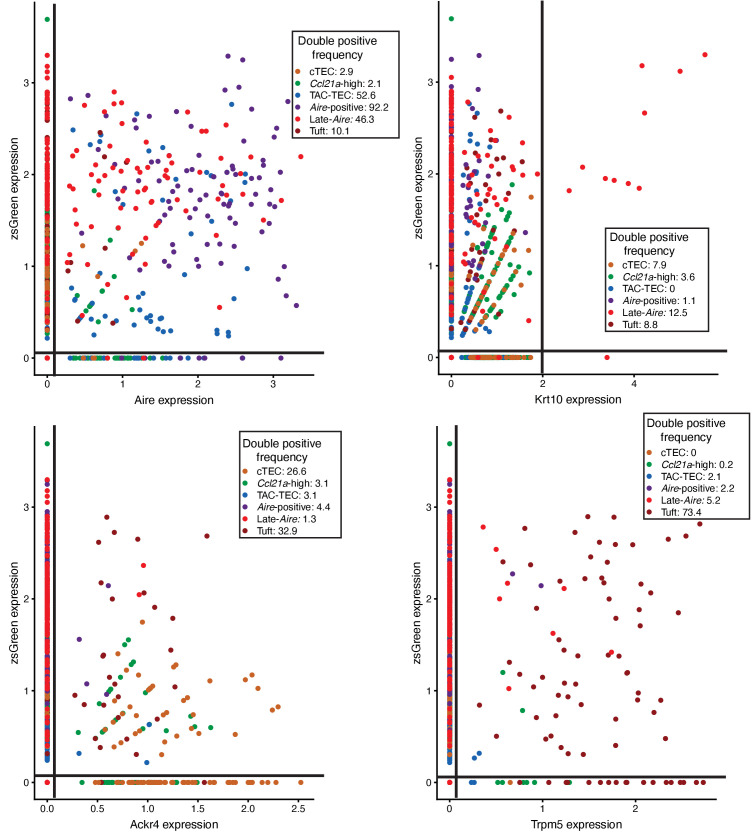
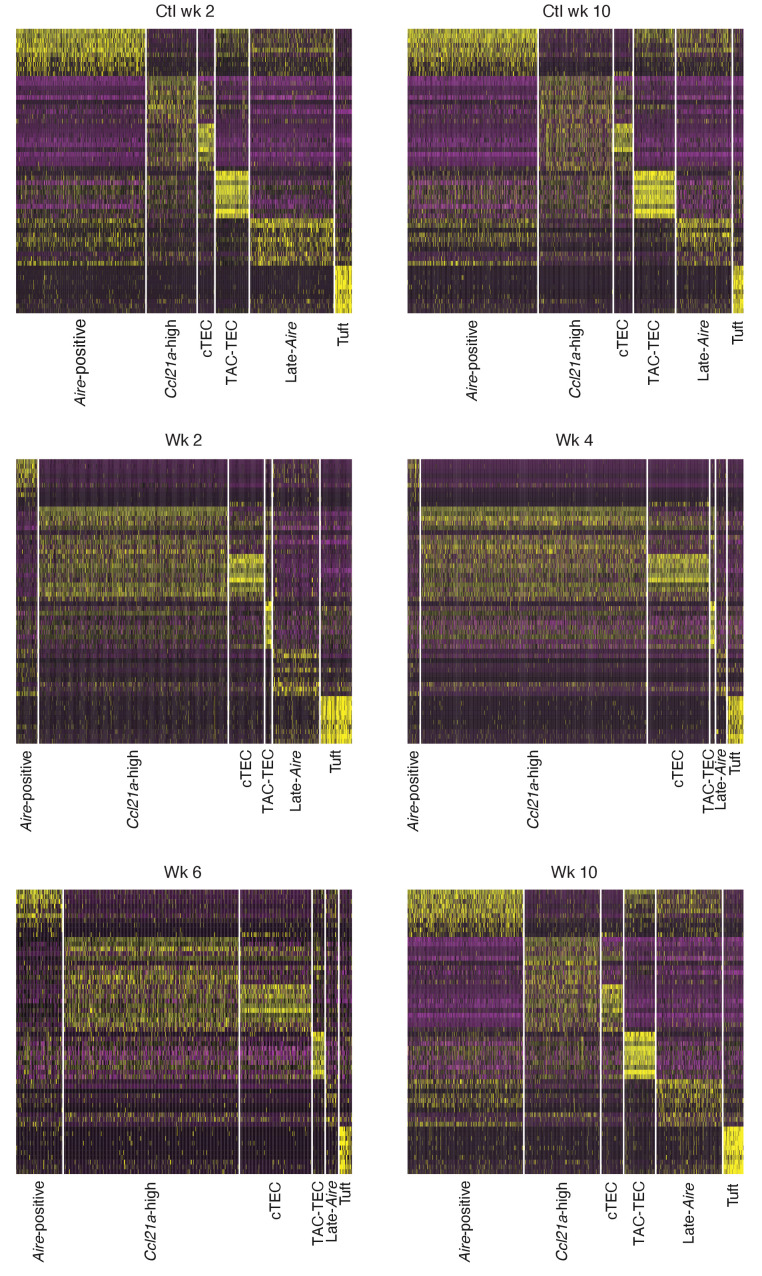
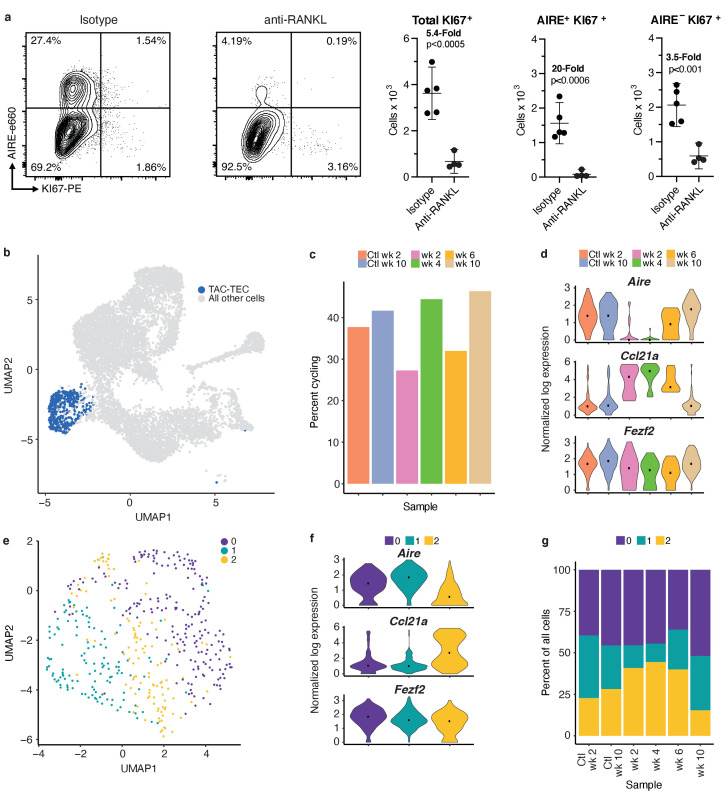
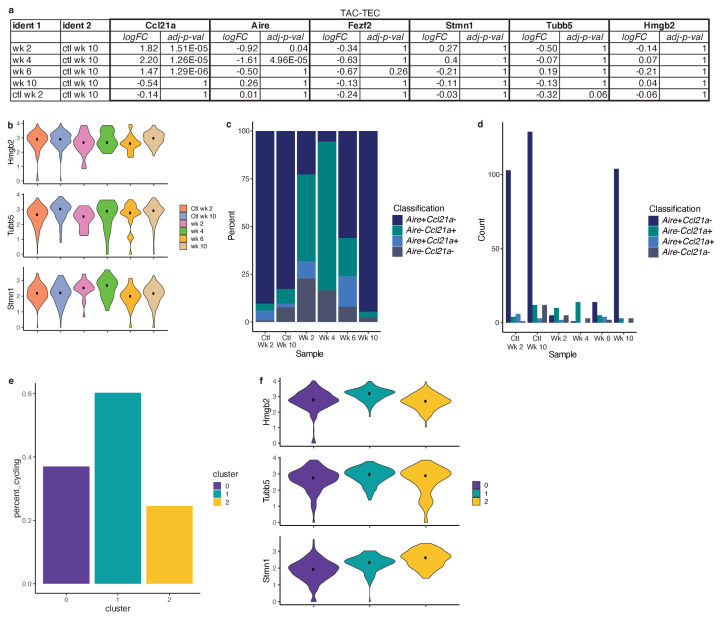
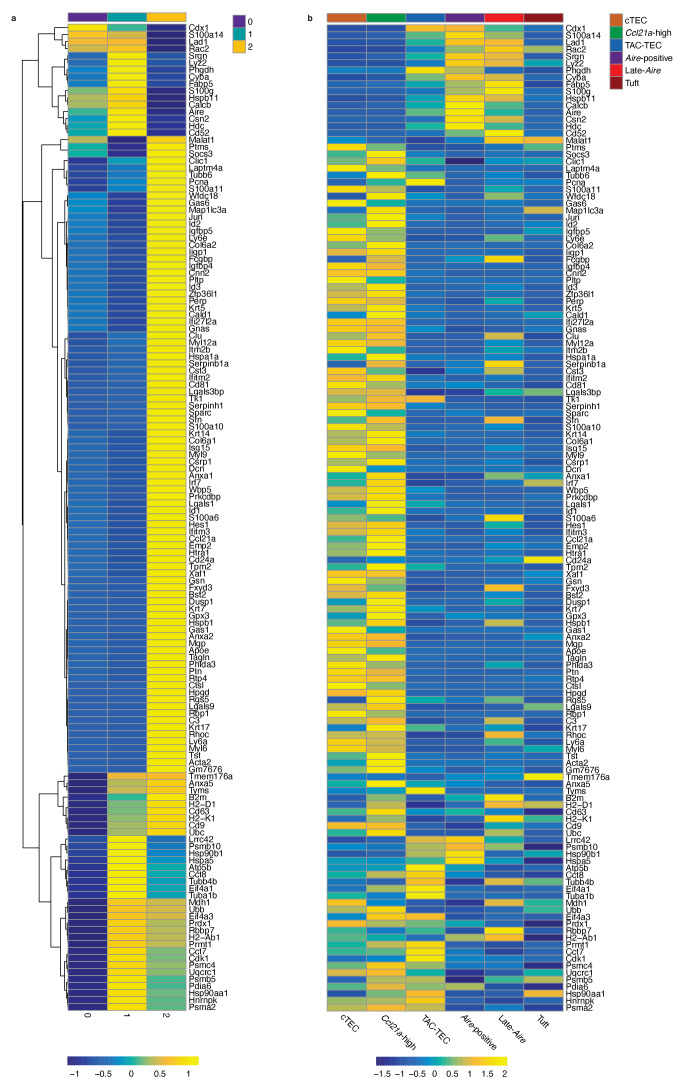
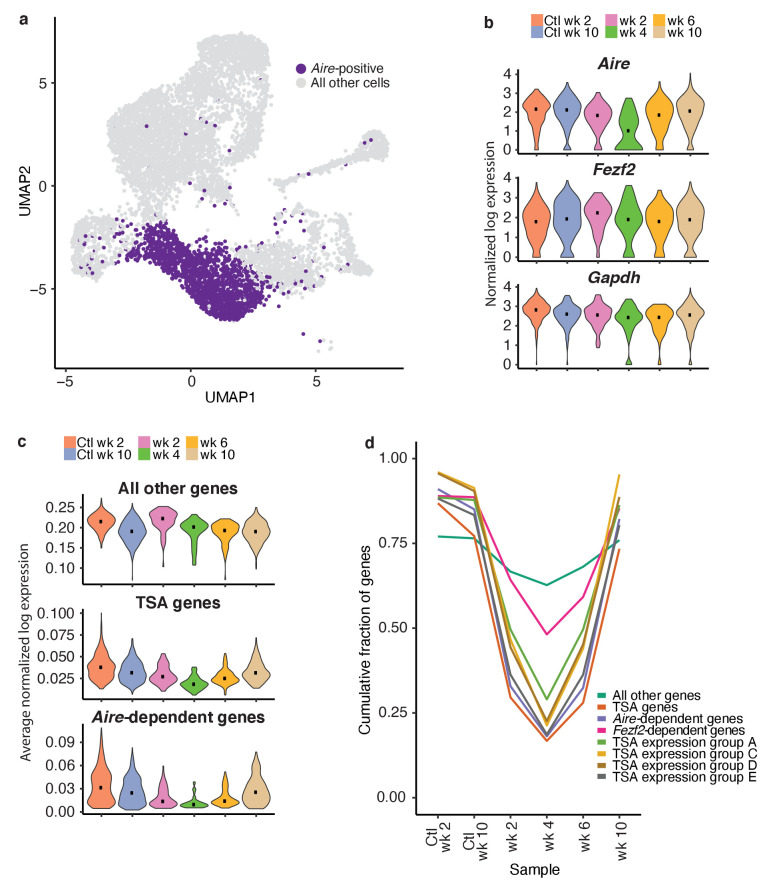
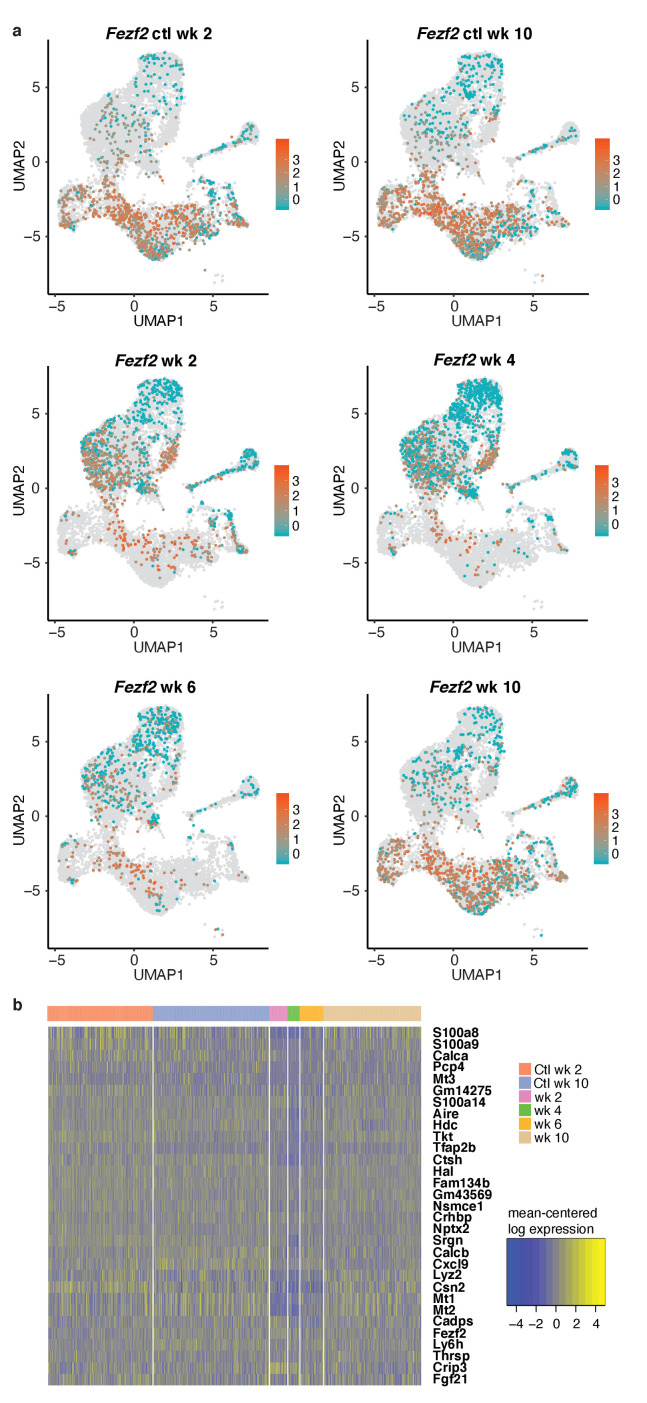
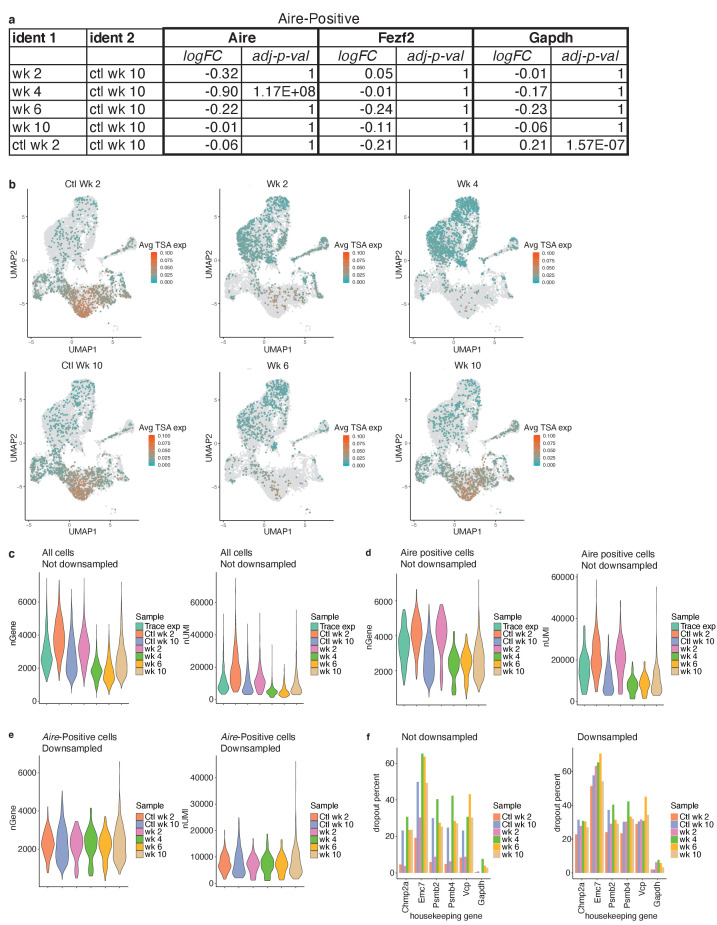
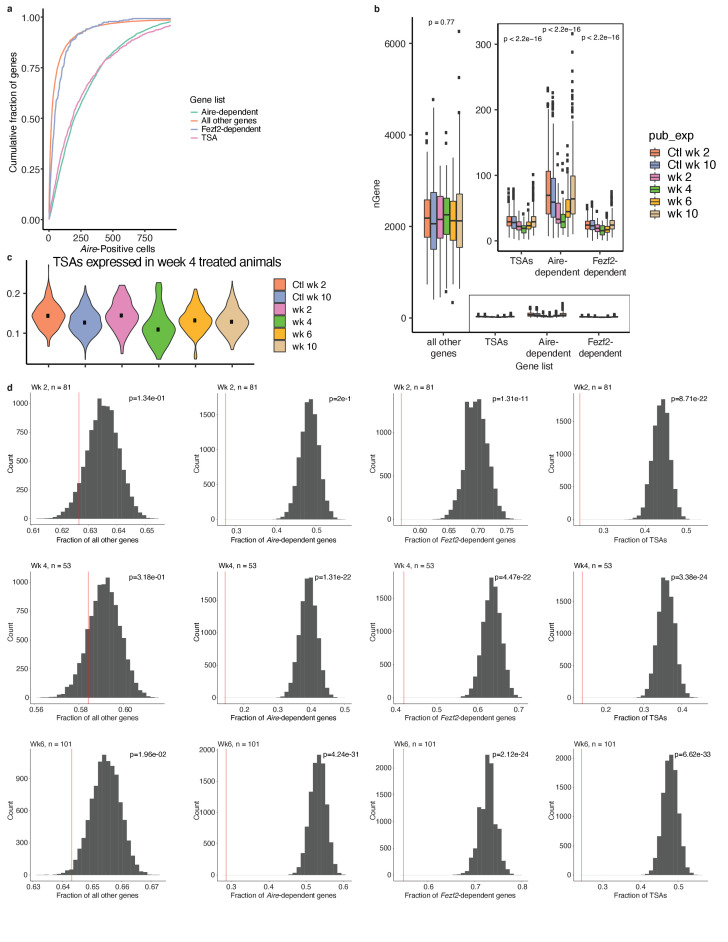
Medullary thymic epithelial cells (mTECs) play a critical role in central immune tolerance by mediating negative selection of autoreactive T cells through the collective expression of the peripheral self-antigen compartment, including tissue-specific antigens (TSAs). Recent work has shown that gene-expression patterns within the mTEC compartment are heterogenous and include multiple differentiated cell states. To further define mTEC development and medullary epithelial lineage relationships, we combined lineage tracing and recovery from transient in vivo mTEC ablation with single-cell RNA-sequencing in Mus musculus. The combination of bioinformatic and experimental approaches revealed a non-stem transit-amplifying population of cycling mTECs that preceded Aire expression. We propose a branching model of mTEC development wherein a heterogeneous pool of transit-amplifying cells gives rise to Aire- and Ccl21a-expressing mTEC subsets. We further use experimental techniques to show that within the Aire-expressing developmental branch, TSA expression peaked as Aire expression decreased, implying Aire expression must be established before TSA expression can occur. Collectively, these data provide a roadmap of mTEC development and demonstrate the power of combinatorial approaches leveraging both in vivo models and high-dimensional datasets.
Keywords: genetics; genomics; immune system; immunology; inflammation; medullary thymic epithelial cell; mouse; single-cell transcriptomics.
Plain language summary
Specialized cells in the immune system known as T cells protect the body from infection by destroying disease-causing microbes, such as bacteria or viruses. T cells use proteins on their surface called receptors to stick to infectious microbes and remove them from the body. Some newly developed T-cells, however, contain receptors that recognize and bind to cells that belong in the body. If these faulty T cells are released, they can attack healthy tissues and cause an autoimmune disease. After a new T cell is developed, it gets carried to a gland in the chest known as the thymus. Cells in the thymus called mTECs screen T cells for receptors that may bind to the body’s tissues. mTECs do this by presenting T cells with proteins that are commonly found on the surface of healthy cells in the body. If a T cell recognizes any of these ‘tissue specific proteins’, it is destroyed or given a new role in the body. Some faulty T cells, however, still manage to evade detection. One way to uncover why this might happen is to investigate how mTECs develop. Previous work showed that mTECs transition through various stages before reaching their final form. However, the order in which these events occur remained unclear. To gain a better understanding of these developmental steps, Wells, Miller et al. extracted mTECs from the thymus of mice and analyzed the genetic make-up of individual cells. This uncovered a missing link in mTEC development: a new type of cell that is the immediate predecessor of the final mTEC. These ‘predecessor’ cells were actively growing, highlighting that mTECs can be constantly generated in the body. By probing the genes that generate tissue-specific proteins in mTECs, Wells, Miller et al. revealed that these proteins were only produced for short periods and in the late stages of mTEC development. These findings contribute to our understanding of how mTECs develop to screen T cells. Mapping these developmental stages will make it easier to identify when faulty T cells are able to evade mTECs. This will lead to earlier detection of autoimmune diseases which could result in better treatments.
© 2020, Wells et al.
Conflict of interest statement
KW, CM, AG, WW, JP, MA, LS No competing interests declared
Figures
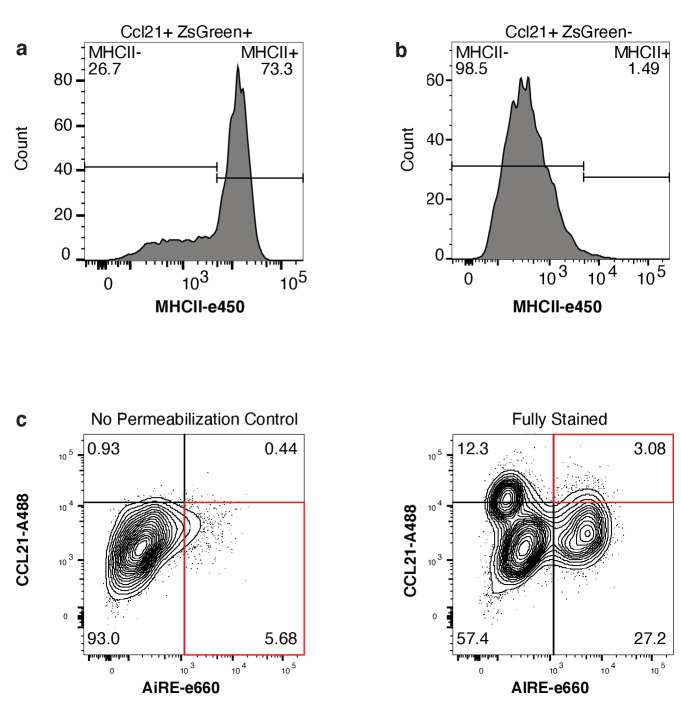
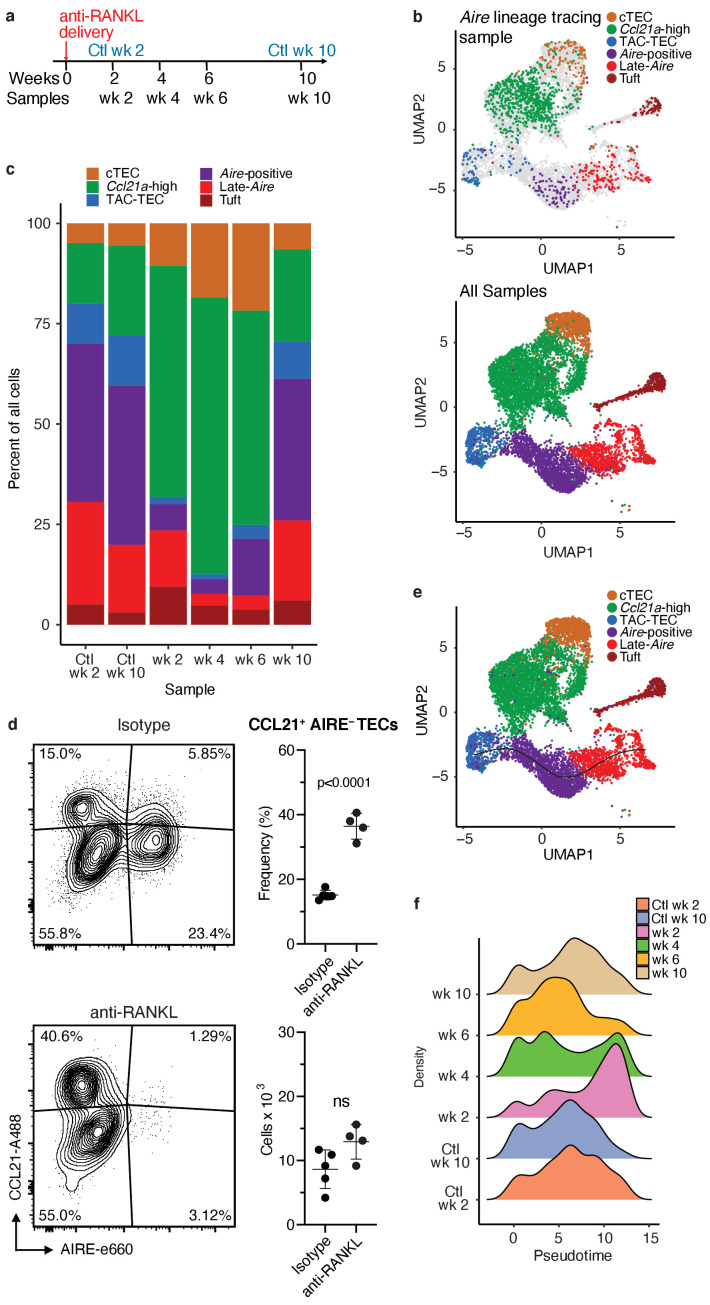
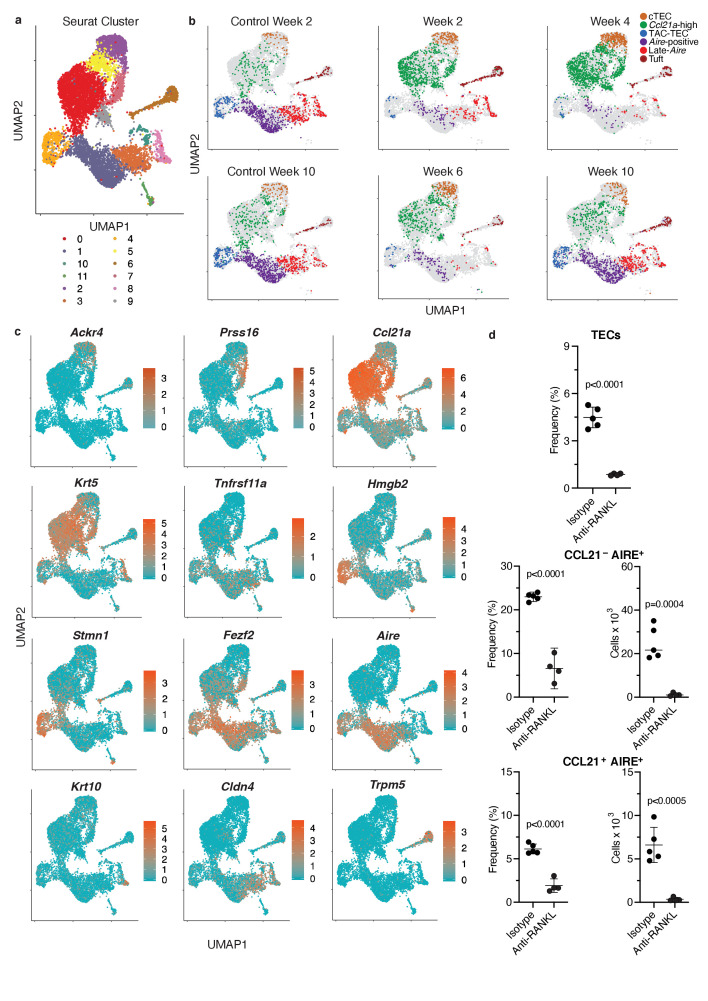
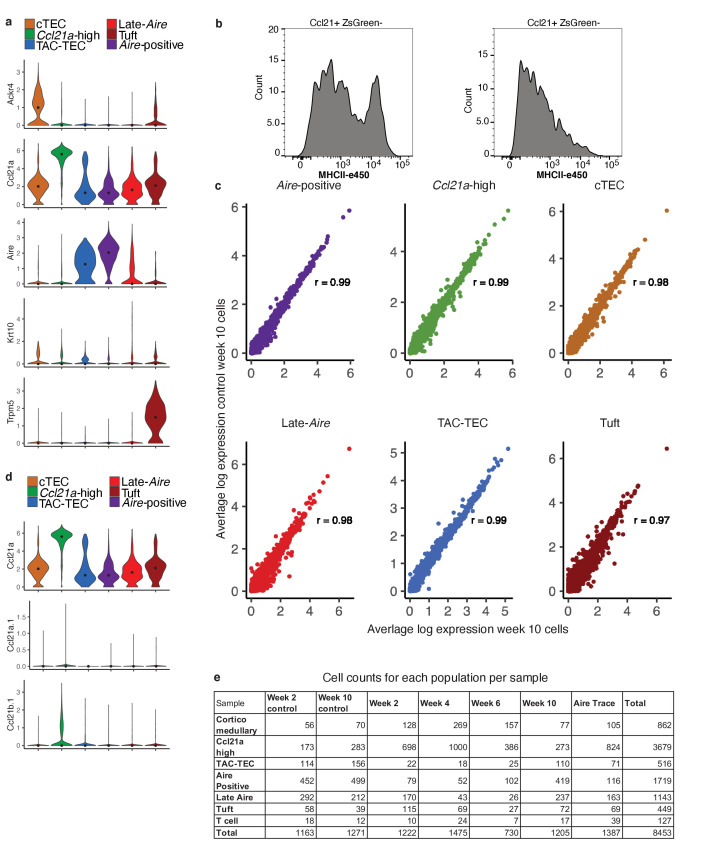
References
-
- Akiyama N, Shinzawa M, Miyauchi M, Yanai H, Tateishi R, Shimo Y, Ohshima D, Matsuo K, Sasaki I, Hoshino K, Wu G, Yagi S, Inoue J, Kaisho T, Akiyama T. Limitation of immune tolerance-inducing thymic epithelial cell development by Spi-B-mediated negative feedback regulation. Journal of Experimental Medicine. 2014;211:2425–2438. doi: 10.1084/jem.20141207. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
