

ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress
- PMID: 33229544
- PMCID: PMC7733819
- DOI: 10.1073/pnas.1922342117
ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress
Abstract
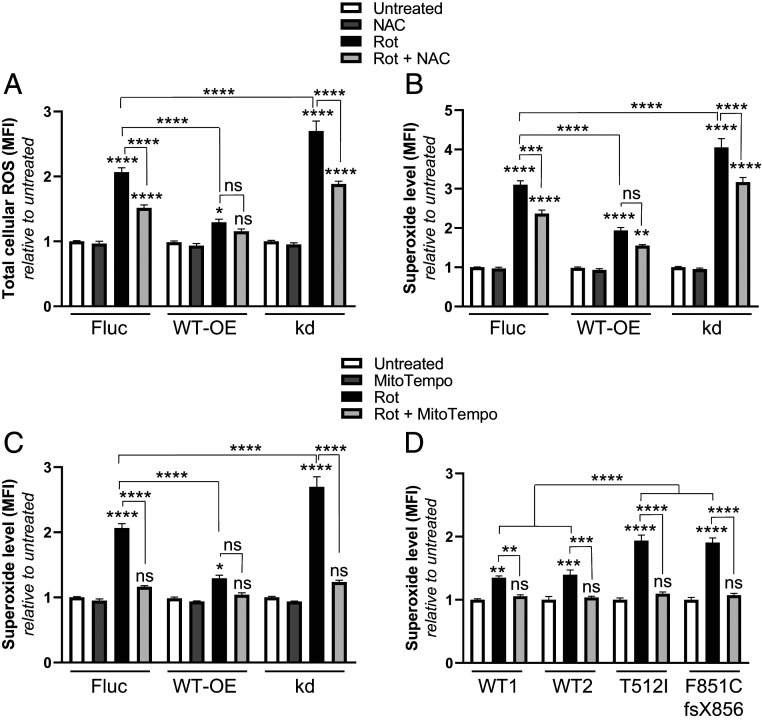
Recessive loss-of-function mutations in ATP13A2 (PARK9) are associated with a spectrum of neurodegenerative disorders, including Parkinson's disease (PD). We recently revealed that the late endo-lysosomal transporter ATP13A2 pumps polyamines like spermine into the cytosol, whereas ATP13A2 dysfunction causes lysosomal polyamine accumulation and rupture. Here, we investigate how ATP13A2 provides protection against mitochondrial toxins such as rotenone, an environmental PD risk factor. Rotenone promoted mitochondrial-generated superoxide (MitoROS), which was exacerbated by ATP13A2 deficiency in SH-SY5Y cells and patient-derived fibroblasts, disturbing mitochondrial functionality and inducing toxicity and cell death. Moreover, ATP13A2 knockdown induced an ATF4-CHOP-dependent stress response following rotenone exposure. MitoROS and ATF4-CHOP were blocked by MitoTEMPO, a mitochondrial antioxidant, suggesting that the impact of ATP13A2 on MitoROS may relate to the antioxidant properties of spermine. Pharmacological inhibition of intracellular polyamine synthesis with α-difluoromethylornithine (DFMO) also increased MitoROS and ATF4 when ATP13A2 was deficient. The polyamine transport activity of ATP13A2 was required for lowering rotenone/DFMO-induced MitoROS, whereas exogenous spermine quenched rotenone-induced MitoROS via ATP13A2. Interestingly, fluorescently labeled spermine uptake in the mitochondria dropped as a consequence of ATP13A2 transport deficiency. Our cellular observations were recapitulated in vivo, in a Caenorhabditis elegans strain deficient in the ATP13A2 ortholog catp-6 These animals exhibited a basal elevated MitoROS level, mitochondrial dysfunction, and enhanced stress response regulated by atfs-1, the C. elegans ortholog of ATF4, causing hypersensitivity to rotenone, which was reversible with MitoTEMPO. Together, our study reveals a conserved cell protective pathway that counters mitochondrial oxidative stress via ATP13A2-mediated lysosomal spermine export.
Keywords: P5B-type ATPase; antioxidant; mitochondria; neurodegeneration; polyamine transport.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
Competing interest statement: Patent WO-2018002350-A1 of KU Leuven describes methods for detecting compounds with therapeutic use that target ATP13A2 or related isoforms using biological material and assays described in the current manuscript. A second patent of KU Leuven describing ATP13A2 cell models described in this manuscript has also been filed.
Figures





References
-
- Ramirez A., et al. , Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191 (2006). - PubMed
-
- Di Fonzo A. et al.; Italian Parkinson Genetics Network , ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562 (2007). - PubMed
-
- Lin C. H., et al. , Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology 71, 1727–1732 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
