

Corticotropin-releasing hormone (CRH) alters mitochondrial morphology and function by activating the NF-kB-DRP1 axis in hippocampal neurons
- PMID: 33230105
- PMCID: PMC7683554
- DOI: 10.1038/s41419-020-03204-3
Corticotropin-releasing hormone (CRH) alters mitochondrial morphology and function by activating the NF-kB-DRP1 axis in hippocampal neurons
Abstract
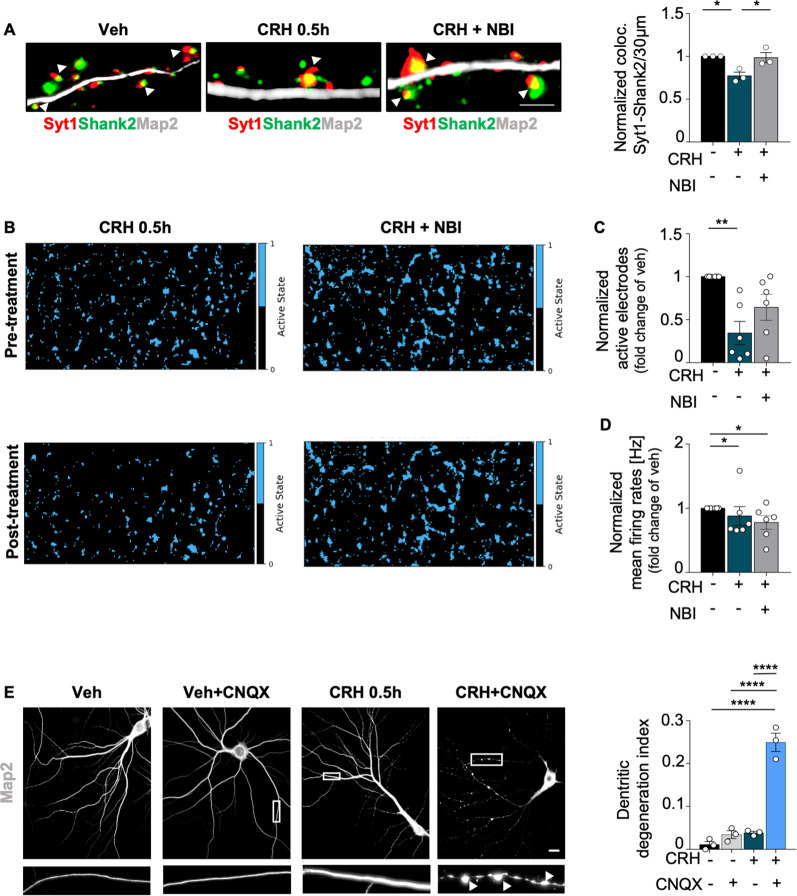
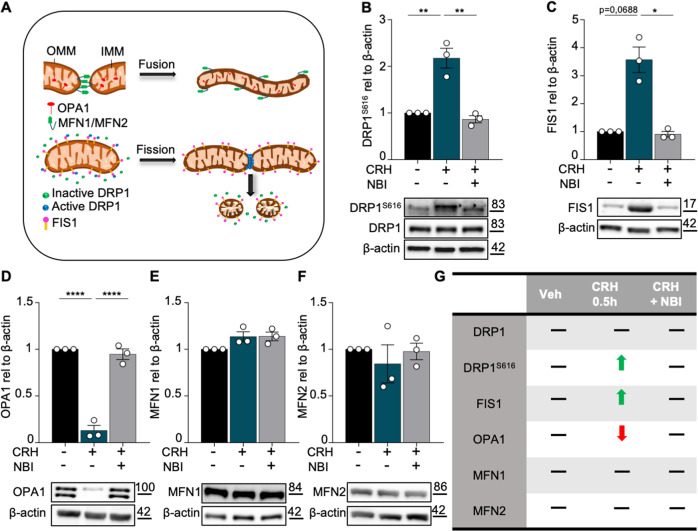
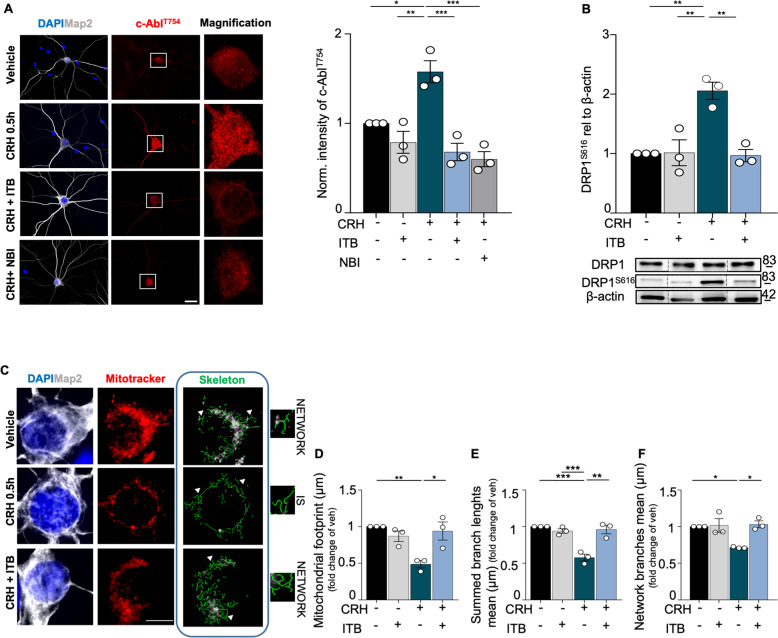
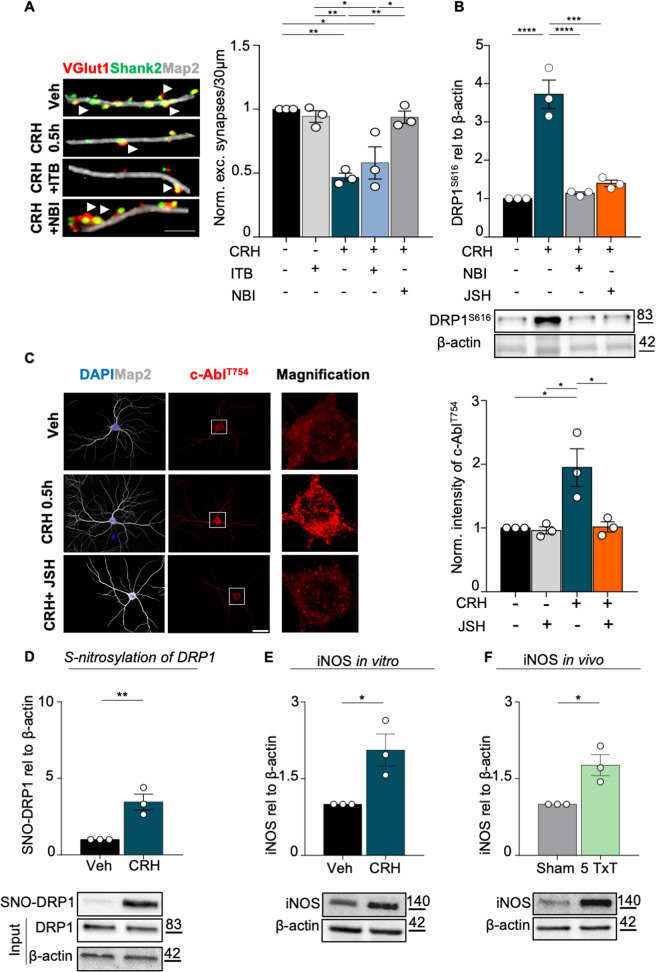
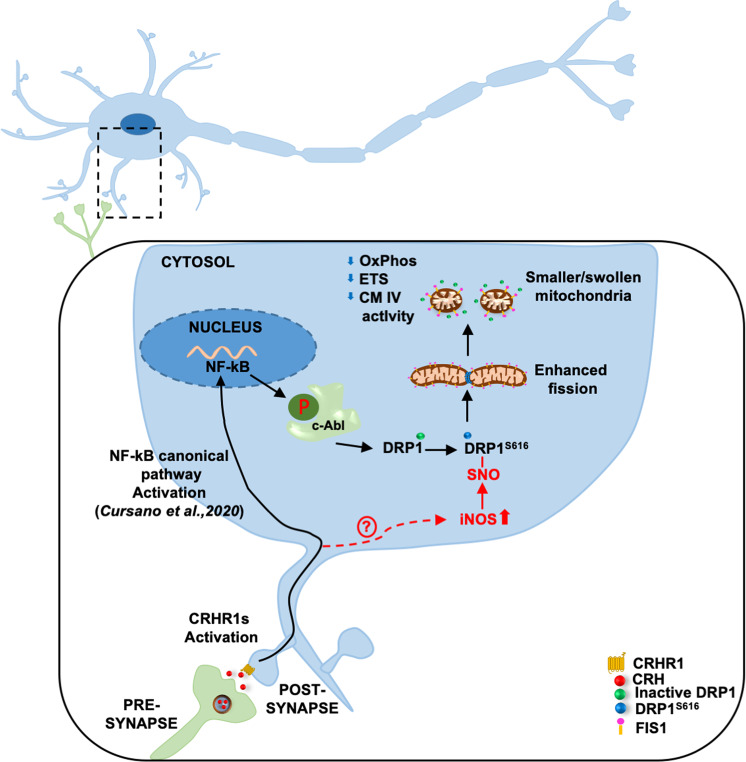
Neuronal stress-adaptation combines multiple molecular responses. We have previously reported that thorax trauma induces a transient loss of hippocampal excitatory synapses mediated by the local release of the stress-related hormone corticotropin-releasing hormone (CRH). Since a physiological synaptic activity relies also on mitochondrial functionality, we investigated the direct involvement of mitochondria in the (mal)-adaptive changes induced by the activation of neuronal CRH receptors 1 (CRHR1). We observed, in vivo and in vitro, a significant shift of mitochondrial dynamics towards fission, which correlated with increased swollen mitochondria and aberrant cristae. These morphological changes, which are associated with increased NF-kB activity and nitric oxide concentrations, correlated with a pronounced reduction of mitochondrial activity. However, ATP availability was unaltered, suggesting that neurons maintain a physiological energy metabolism to preserve them from apoptosis under CRH exposure. Our findings demonstrate that stress-induced CRHR1 activation leads to strong, but reversible, modifications of mitochondrial dynamics and morphology. These alterations are accompanied by bioenergetic defects and the reduction of neuronal activity, which are linked to increased intracellular oxidative stress, and to the activation of the NF-kB/c-Abl/DRP1 axis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
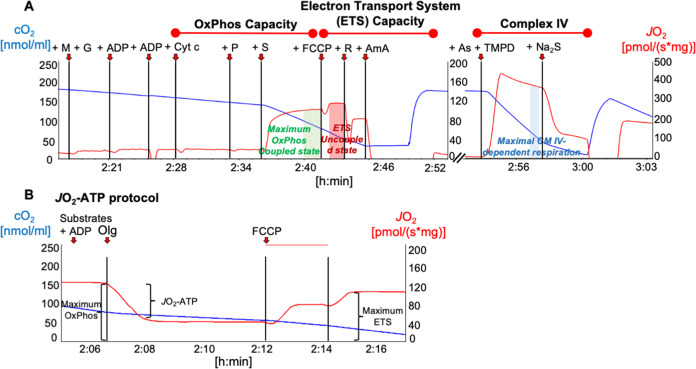
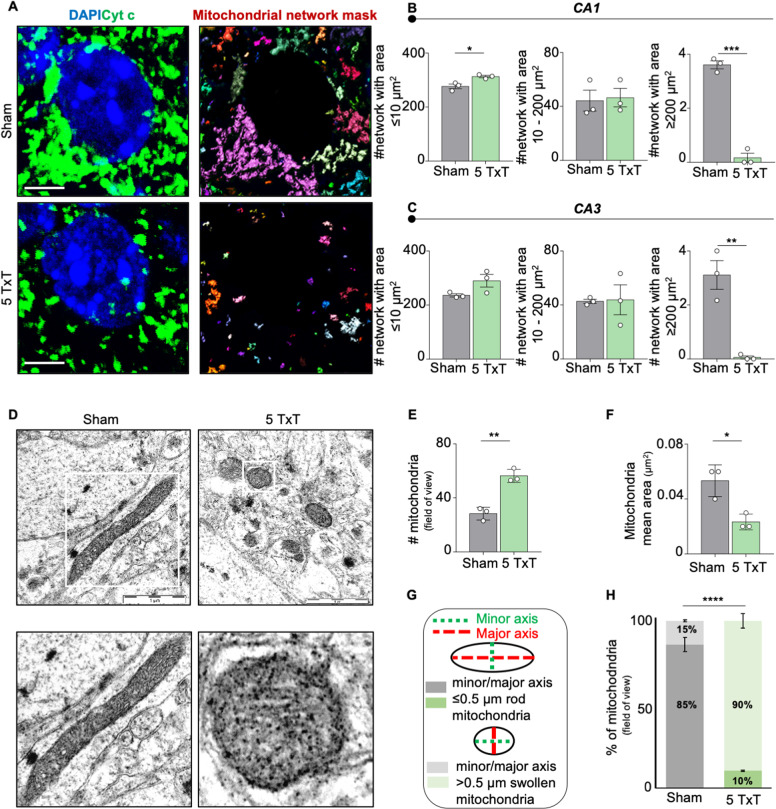
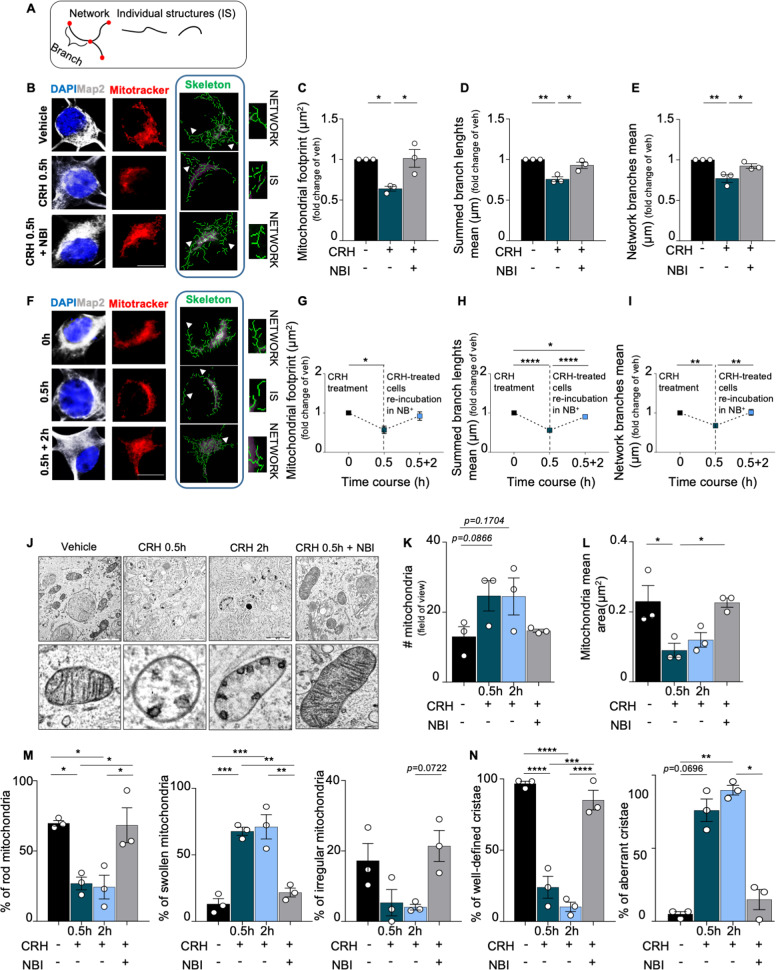
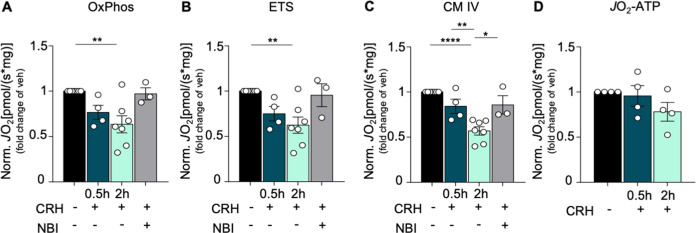
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
