

Epidemiology and biology of a herpesvirus in rabies endemic vampire bat populations
- PMID: 33230120
- PMCID: PMC7683562
- DOI: 10.1038/s41467-020-19832-4
Epidemiology and biology of a herpesvirus in rabies endemic vampire bat populations
Abstract
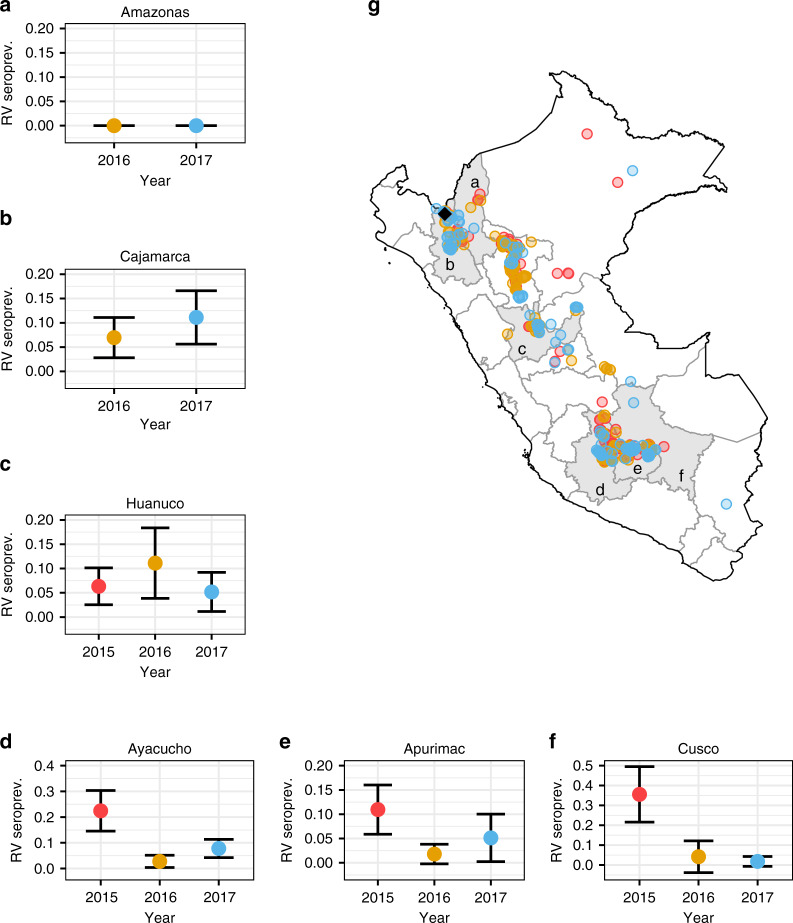
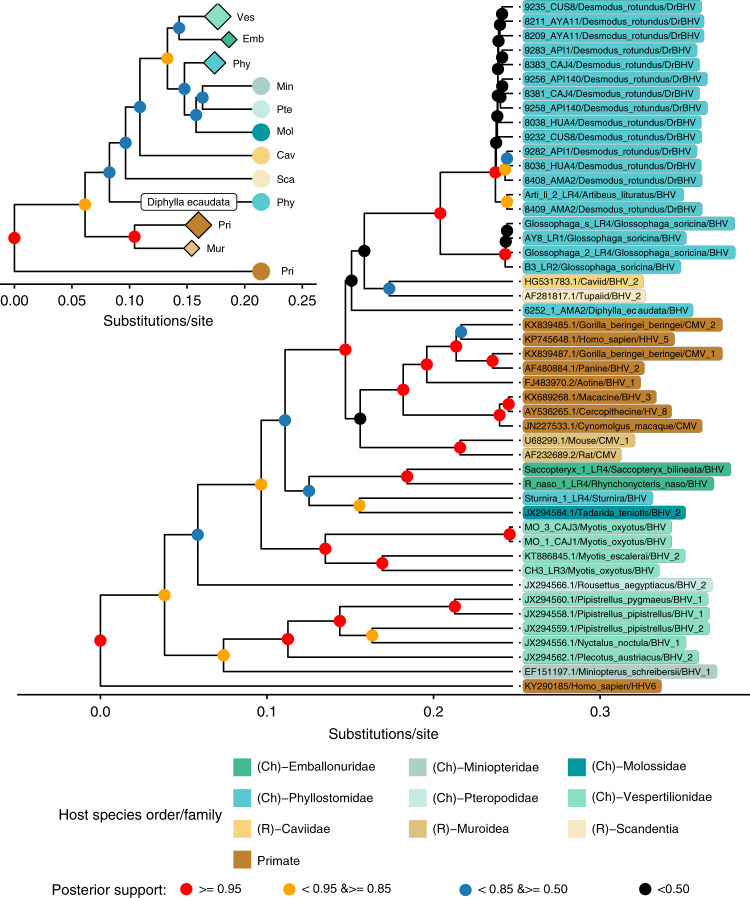
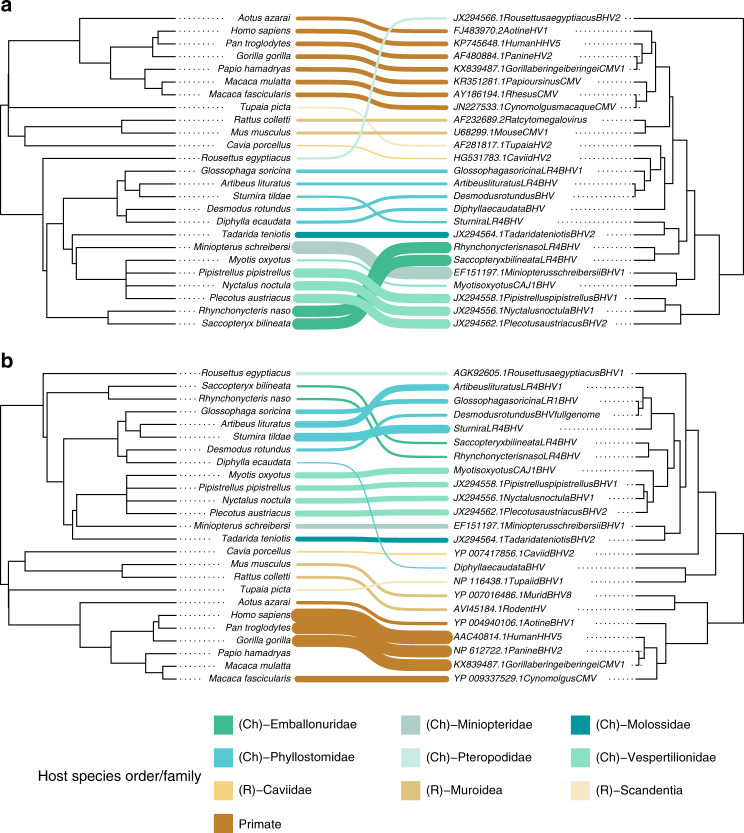
Rabies is a viral zoonosis transmitted by vampire bats across Latin America. Substantial public health and agricultural burdens remain, despite decades of bats culls and livestock vaccinations. Virally vectored vaccines that spread autonomously through bat populations are a theoretically appealing solution to managing rabies in its reservoir host. We investigate the biological and epidemiological suitability of a vampire bat betaherpesvirus (DrBHV) to act as a vaccine vector. In 25 sites across Peru with serological and/or molecular evidence of rabies circulation, DrBHV infects 80-100% of bats, suggesting potential for high population-level vaccine coverage. Phylogenetic analysis reveals host specificity within neotropical bats, limiting risks to non-target species. Finally, deep sequencing illustrates DrBHV super-infections in individual bats, implying that DrBHV-vectored vaccines might invade despite the highly prevalent wild-type virus. These results indicate DrBHV as a promising candidate vector for a transmissible rabies vaccine, and provide a framework to discover and evaluate candidate viral vectors for vaccines against bat-borne zoonoses.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Organization, W. H. WHO Expert Consultation on Rabies: Second Report (World Health Organization, 2013). - PubMed
-
- Schneider MC, et al. Rabies transmitted by vampire bats to humans: an emerging zoonotic disease in Latin America? Rev. Panam. Salud Publica Pan Am. J. Public Health. 2009;25:260–269. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
